 |
Clinical Image
Rare case of Pyle’s metaphyseal dysplasia presenting in an adult
1 5th Year Resident, Department of Orthopedic Surgery and Traumatology, Hospital Espírito Santo de Évora, Évora, Portugal
2 Specialist Surgeon, Department of Orthopedic Surgery and Traumatology, Hospital Espírito Santo de Évora, Évora, Portugal
3 4th Year Resident, Department of Orthopedic Surgery and Traumatology, Hospital Espírito Santo de Évora, Évora, Portugal
Address correspondence to:
Jorge Sena
Rua Ruy Luís Gomes n293, 2925-689 Azeitão-Setúbal,
Portugal
Message to Corresponding Author
Article ID: 101088Z01JS2020
Access full text article on other devices

Access PDF of article on other devices


How to cite this article
Sena J, Xavier G, Sampaio J, Nóbrega J. Rare case of Pyle’s metaphyseal dysplasia presenting in an adult. Int J Case Rep Images 2020;11:101088Z01JS2020.ABSTRACT
No Abstract
Keywords: Bone morphogenetic protein, Dysplasia, Erlenmeyer flask, Niemann–Pick’s disease, Pyle’s metaphyseal dysplasia
Case Report
A 75-year-old woman presented at our institution with gonalgia and consequently functional limitation of the right knee, for several weeks. At physical examination, besides the bilateral and symmetrical enlargement of the knees, it was evident the marked bilateral genu valgum and wasting of legs. The X-ray showed bizarre bone changes of the femur and tibia. There was no family history of similar changes.
Besides these changes there was no other medical history issue, and the patient was not under any specific medication, besides pain killers.
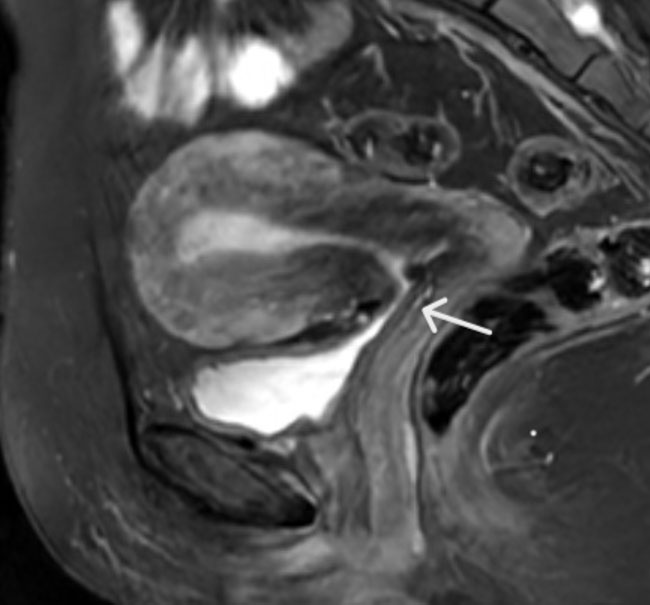
For further investigation of the patient complains we required additional X-rays of other anatomical regions in skeletal survey. In the femora, the changes were most marked distally where gross “Erlenmeyer flask” deformities were evident (Figure 1). This flaring of the femur extended proximal to the mid-femoral diaphysis where endosteal cortical thickening was present. Tibiae and fibulae were expanded with mid-diaphyseal cortical thickening and lateral bowing of the tibiae (Figure 2). Other X-rays also revealed undertubulation of the superior limbs (Figure 3 and Figure 4) and pelvic changes (Figure 5).





Discussion
Pyle’s metaphyseal dysplasia (PMD) is an extremely uncommon and rare genetic skeletal disorder, with fewer than 30 cases reported worldwide, characterized by a benign course of irregular development of long bones, and an autosomal recessive genetic pattern of inheritance [1],[2],[3]. This disease was named after the American orthopedic surgeon, Edwin Pyle, who first reported the disease in 1931. Clinical manifestations contrast with radiological appearances of gross metaphyseal undermodeling which widening of long bones with marked cortical changes (Erlenmeyer-flask deformity) especially around the knee. Proximal humerus and distal radius and ulna eventually show undertubulation changes. Other less marked changes may be seen in other distal long bones, distal metacarpals and proximal phalanges. Spinal involvement varies from no changes at all to a moderate platyspondyly or biconcave lens appearance of the vertebral bodies [1],[4]. Homozygous mutations in secreted frizzled-related protein 4 (SFRP4) gene have been related to this condition. Secreted frizzled-related protein 4 gene (OMIM 606570) produces a protein that acts as an antagonist to the WNT/frizzled pathway when bound to the WNT ligands or to the frizzled receptors. These mutations (stop mutations) have been reported to affect the downregulation of SFRP on WNT and bone morphogenetic protein (BMP) signaling pathways, which may cause abnormal thinning and osteoporosis of the bone cortex [5].
The differential diagnosis for PMD includes other craniotubular bone dysplasias, such as craniometaphyseal dysplasia, craniodiaphyseal dysplasia, osteopetrosis, Niemann–Pick’s disease, Gaucher disease, and thalassaemia. Pyle’s disease is often confused with craniometaphyseal dysplasia. In 1970, Gorlin et al. described craniometaphyseal dysplasia as a distinct entity separate from Pyle’s disease. While PMD has gross widening of metaphyses with mild cranial sclerosis, other craniometaphyseal dysplasias have severe craniofacial or greater cranial sclerosis with mild metaphyseal changes [3],[4].
Absence of cranial involvement excludes first three differential diagnoses in our patient. Moreover, our patient did not have anemia, jaundice, hepatosplenomegaly, increased bone density or any history of blood transfusion ruling out the latter four possibilities also.
Clinical–radiological dissociation associated with characteristic imaging deformities was consistent with the diagnosis of Pyle’s syndrome. The vast majority of PMD cases are asymptomatic and their diagnosis is predominantly the result of an accidental finding observed on radiographs requested for other reasons. Treatment is not required for this condition, being reserved for the eventuality of a fracture or for arthrosis resulting from the genu valgum that requires surgical intervention.
Conclusion
This case is presented due to the rarity and typical radiological presentation, highlighting the clinical signs, X-rays, and differential diagnosis. A physician aware of this group of pathologies will know how to make the diagnosis thus avoiding more aggressive or costly diagnostic approaches [computed tomography (CT), magnetic resonance imaging (MRI), biopsy, and genetic studies] and the anxiety/alarmism of the patient and family in the face of diagnostic uncertainty. After diagnosis, the patient was proposed for total right knee arthroplasty due to pain complaints and grade IV gonarthrosis, which she refused, alternatively choosing conservative treatment.
REFERENCES
1.
Soares DX, Almeida AM, Barreto AR, et al. Pyle disease (metaphyseal dysplasia) presenting in two adult sisters. Radiol Case Rep 2016;11(4):405–10. [CrossRef]
[Pubmed]

2.
Adejimi OA. Pyle-type metaphyseal dysplasia. Appl Radiol 2009;38:40A–C.
3.
Wonkam A, Makubalo N, Roberts T, Chetty M. Pyle metaphyseal dysplasia in an African child: Case report and review of the literature. S Afr Med J 2016;106(6 Suppl 1):S110–3. [CrossRef]
[Pubmed]
4.
Galada C, Shah H, Shukla A, Girisha KM. A novel sequence variant in SFRP4 causing Pyle disease. J Hum Genet 2017;62(5):575–6. [CrossRef]
[Pubmed]
5.
Gupta N, Kabra M, Das CJ, Gupta AK. Pyle metaphyseal dysplasia. Indian Pediatr 2008;45(4):323–5.
[Pubmed]
SUPPORTING INFORMATION
Author Contributions
Jorge Sena - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Gabriel Xavier - Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
João Sampaio - Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
João Nóbrega - Acquisition of data, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Guarantor of SubmissionThe corresponding author is the guarantor of submission.
Source of SupportNone
Consent StatementWritten informed consent was obtained from the patient for publication of this article.
Data AvailabilityAll relevant data are within the paper and its Supporting Information files.
Conflict of InterestAuthors declare no conflict of interest.
Copyright© 2020 2020 Jorge Sena et al.. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}