
|
 |
|
Case Report
| ||||||
| Treatment of Epstein–Barr virus induced hemophagocytic lymphohistiocytosis with anti-CD20 therapy: A two-year follow-up | ||||||
| Bijit Kumar Kundu1, Deepak Rath2 | ||||||
|
1MD, Associate Professor, Rheumatology Clinic, Department of Medicine, PGIMER & Dr RML Hospital, New Delhi, India 2MD, Postdoctoral Trainee, Department of Rheumatology, IPGMER & SSKM Hospital, Kolkata, India | ||||||
| ||||||
|
[Abstract HTML]
[Full Text HTML]
[Full Text PDF]
[Print This Article] [Similar article in Pumed] [Similar article in Google Scholar] |


| How to cite this article |
| Kundu BK, Rath D. Treatment of Epstein–Barr virus induced hemophagocytic lymphohistiocytosis with anti-CD20 therapy: A two-year follow-up. Int J Case Rep Images 2018;9(1):25–32. |
|
ABSTRACT
| ||||||
|
Introduction: Hemophagocytic lymphohistiocytosis (HLH) is well known to the rheumatologist as a devastating complication of a rheumatologic disease, being one of the few rheumatologic emergencies. Its treatment is usually started on the basis of presumption without waiting for confirmatory evidence. The diagnosis of HLH is usually missed when it appears in any of its many and varied chronic manifestation. It can be a great mimic with symptoms pertaining to any organ system of the human body. Once diagnosed it is imperative to find the trigger in order to prevent recurrences. Keywords: Epstein–Barr virus, Hemophagocytic lymphohistiocytosis, Panniculitis, Rituximab | ||||||
|
INTRODUCTION
| ||||||
|
Hemophagocytic lymphohistiocytosis (HLH) by itself as well as when occurring secondary to autoimmune diseases when it is called macrophage activation syndrome (MAS); is a devastating event with the patient toxic, having high fever and cytopenias; making it difficult to differentiate it from septicemia. What is however not well known is that HLH can have a low grade, chronic presentation with varied organ specific including dermatological manifestations [1]. This variant of HLH is usually caused by Epstein–Barr virus (EBV) and it is important to not only treat the HLH, but also the accompanying EBV, which is the antigenic trigger and drive leading to HLH [2]. We present a case where a middle-aged lady who had recurrent, slowly growing and resolving nodular swellings with atrophic sequelae on different parts of the body, diagnosed as panniculitis on histopathology studies was found to actually have HLH and was treated with anti-CD20 therapy (Rituximab, RTX) in addition to the treatment of HLH. | ||||||
|
CASE REPORT
| ||||||
|
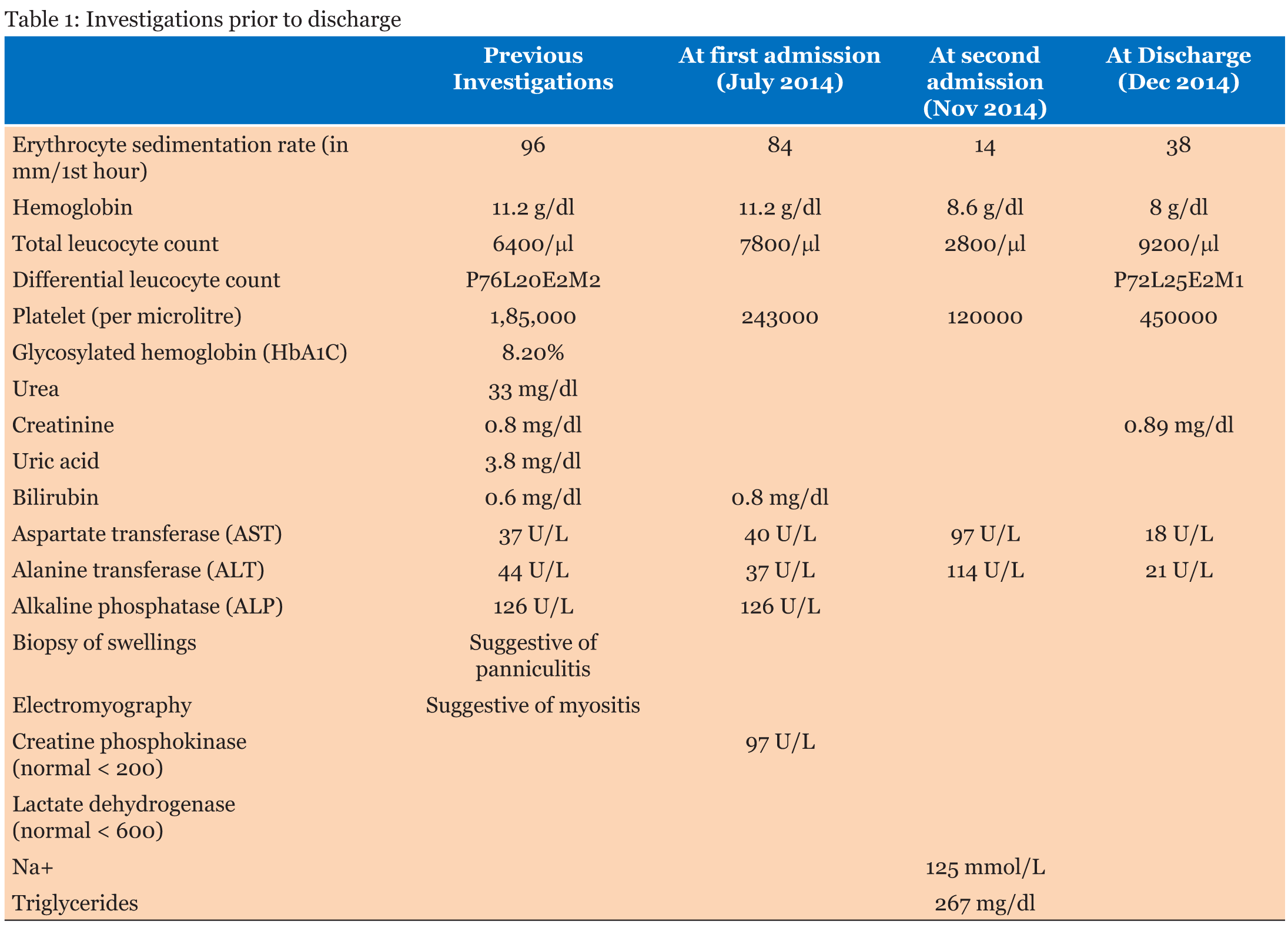
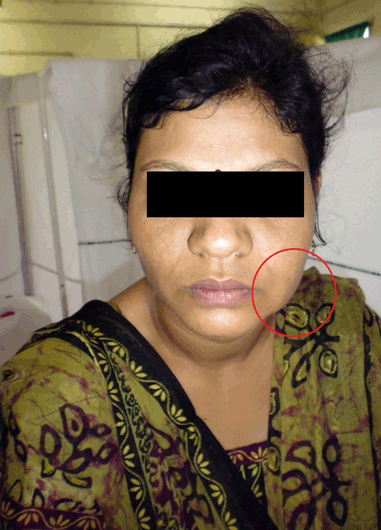
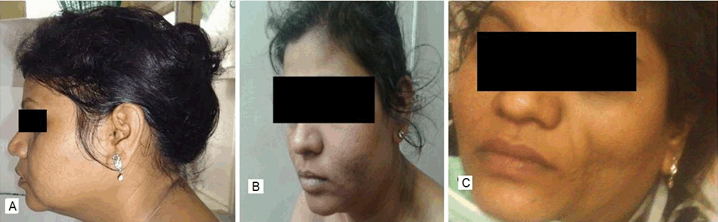
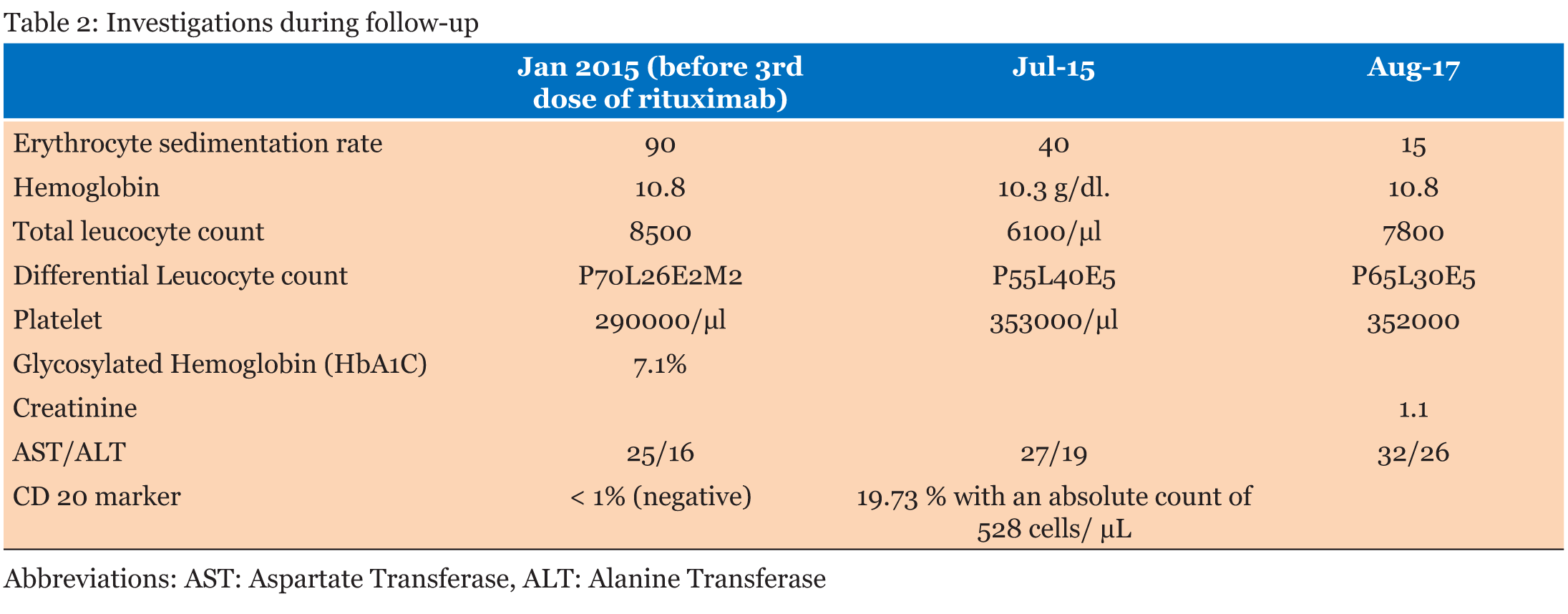
A 37-year-old female was referred to the rheumatology Clinic of our hospital in July 2014 with history of recurrent nodular swellings over face, arms, shoulders, abdomen, and legs for the past 7–8 years and a diagnosis of myositis on electromyography. Review of the history revealed that the swellings had started appearing around eight years back. They would start as a small nodule, gradually increasing in size over the next 1–2 months, reaching a maximum size of about 5 cm and would resolve by itself over another period of 2–3 months leaving behind localized atrophy at the site of the swelling without any residual pigmentary changes. Signs of inflammation (both systemic and local) were conspicuous by their absence except mild warmth to touch. She had 1 or 2 such episodes a year, and at any point of time 1 or 2 swellings at most. There would be intervening period of months without appearance of any new swelling. The swelling had been many times diagnosed as abscesses and treated with antibiotics. The patient had prior involvement of the supraclavicular region, upper arm, back, abdomen and forearm regions which had given her a muscular appearance (Figure 1). Around three years back the patient had developed diabetes and was under treatment with oral anti-diabetic agents (OADs) [gliclazide 80 mg once a day, metformin 500 mg three times a day] but control of diabetes was poor. There was no past history of tuberculosis in self or contact with a patient of tuberculosis or leprosy. She had been suggested to use insulin but never had utilized it. There was no history of any other subcutaneous drug injections or illicit drug use. There was no history of arthritis, rash, muscle pain or weakness. There were no features to suggest diagnosis of any systemic connective tissue disorders (CTD). The investigations done prior to visiting us are given in Table 1. At the time of presenting to us, the patient had swellings on the left cheek and right upper abdomen region which had been present for the last two months. On examination, the patient was a middle-aged female of average built and nutrition with a muscular appearance. General and systemic examinations were normal except for lipoatrophy over bilateral proximal limbs, shoulders and localized areas of lipoatrophy over the breasts and gluteal region. Local examination of the nodules over the left side of the face and right hypochondrium revealed swellings of 2x3 cm with poorly defined borders, firm in consistency, irregular surface and without any fluctuation (Figure 2). Based on the clinical picture and investigations present the possibilities of nodular sarcoidosis, cutaneous T cell lymphoma and lipoatrophic panniculitis [to be evaluated for cause] were kept. The patient was evaluated in view of the above mentioned differential diagnosis. The laboratory investigations are enumerated in Table 1. At our centre, Chest X-ray (CXR) was normal. Tests for Human Immunodeficiency virus (HIV) I and II, hepatitis B surface antigen (HBsAg) and antibody to hepatitis C virus (anti-HCV antibody) were negative. Ultrasonography of abdomen and pelvis was normal. The contrast enhanced computed tomography (CECT) of chest did not show any mass or hilar lymphadenopathy. Anti-nuclear antibodies (ANA) and antibodies to double-strand deoxyribonucleic acid (dsDNA) were negative. However, the complement levels were raised: C3: 187.4 (75–135 mg/dl); C4: 51.8 (13–45 mg/dL); which is usually not found in autoimmune processes. Immunoglobulin G (IgG) levels were raised: 2251.4 (620–1400 mg/dl) while Immunoglobulin M (IgM) levels were normal: 176.2 (45–250 mg/dl). Fine needle aspiration cytology (FNAC) of the cheek swelling showed only non-specific inflammatory changes and no acid-fast bacilli/granulomas were seen. The thyroid profile of the patient was normal. T3: 95.30 ng/dl (84.6–207.8), T4: 11.87 mcg/dl (5.13–14.06), TSH: 3.34 µIU/mL (0.2–4.2). Muscle biopsy done in view of the earlier EMG report did not show any abnormality. As no definitive diagnosis could be reached at this stage, she was sent back with optimization of anti-diabetic therapy. She was asked to report back in case of any new development. In mid-October 2014, the patient developed fever which was initially low grade but progressed to become high grade and continuous in nature without any response to antipyretics. This was associated with increase in the size of the nodule over the face as described previously. She was prescribed antibiotics locally without any affect. She reported to us in the first week of November 2014 and was admitted. On examination she was found to have temperature of 105°F with a toxic look, pallor and cervical lymphadenopathy. The lymph nodes were of size 1x1 cm to 2x1 cm, multiple, non-tender, discrete and firm in consistency. Systemic examination revealed hepatosplenomegaly. Investigations done at second admission are given in Table 1. The total serum iron 72 (25–170 µg/dl) and transferrin saturation 35.6% (20–55%) were normal. A repeat chest X-ray and ultrasonography were normal. The patient was started on empirical antibiotics and supportive care pending further evaluation. Fine needle aspiration cytology (FNAC) of the cervical lymph nodes was reported to be having reactive changes only. A biopsy of the cheek swelling was reported as panniculitis, but the type could not be commented. The constellation of fever, lymphadenopathy, hepatosplenomegaly, bicytopenia, raised liver enzymes, hyper-triglyceridemia and hyponatremia prompted us to think of HLH and the patient was further worked-up for the same. Retrospective examination of some of her old reports revealed that her investigations had shown bicytopenia and normal ESR during November 2009, but it was not followed up any further then. The serum ferritin levels were markedly elevated [>2000 ng/ml (normal range: 4.6–204)]. But the bone marrow examination did not reveal hemophagocytosis. However, our patient fulfilled 5 of 8 criteria: namely fever, splenomegaly, bicytopenia, hyper-ferritinemia and hypertriglyceridemia needed for the diagnosis of HLH. Hence the diagnosis of HLH was kept, and patient was evaluated further for the underlying cause. Review of the literature revealed that panniculitis can be a presentation of HLH and low grade HLH can occur with Epstein–Barr virus (EBV) infection. Hence EBV serology was sent. In view of the deteriorating condition of the patient, treatment of HLH was started pending the report of EBV serology report. The patient was put on pulse methylprednisolone (1 g once a day for 3 days) followed by dexamethasone 10 mg/m2 (20 mg/day) and cyclosporine 3 mg/kg (150 mg once a day). The patient responded favourably and was afebrile after 12 hours of initiation of methylprednisolone. The swelling over the face also started to decrease (Figure 3). The antibody to Epstein–Barr virus viral-capsid antigen (EBV-VCA) was strongly positive for both IgM and IgG. [EBV IgM 45.30 U/mL (normal range: <8.00) and IgG EBV >200 U/mL (normal <8.00)] Hence based on a positive EBV serology in a patient of fulfilling five of the eight criteria of diagnosis of HLH, a diagnosis of EBV induced HLH (initially indolent, but now full blown) with panniculitis was made and anti-CD20 therapy (rituximab 500 mg (@375 mg/m2)) was also concurrently started. The patient continued to improve with resolution of bicytopenia, raised liver enzymes, and regression of hepatosplenomegaly. The investigations prior to discharge are given in Table 1. The patient received the second dose of rituximab after three weeks. Laboratory reports in January 2015, after eight weeks and two doses of rituximab are given in Table 2. The patient was given total of five doses of rituximab every three weeks. After 16 weeks of the last dose, her laboratory reports showed HbA1C of 7.1% (Table 2). She has been following-up with us regularly since then. Her latest reports dated 30/08/2017 are given in Table 2. As of date she has had no recurrence of the nodules. | ||||||
| ||||||
| ||||||
| ||||||
| ||||||
| ||||||
|
DISCUSSION
| ||||||
|
Hemophagocytic lymphohistiocytosis is of two types, namely primary which includes usually infants and young children with clear familial inheritance or genetic causes and secondary, which affects older children and adults without any family history or genetic causes but having a concurrent condition like infection, malignancy, or rheumatologic disorder. Discovery that the genetic defects can present even in adults, and that infections can be a precipitating factor in such patients have made the designations of ‘primary’ and ‘secondary’ less relevant. Instead, the terms ‘genetic’ and ‘acquired’ HLH have come into vogue reflecting the aetiology more accurately [2]. The causes of acquired HLH can thus be divided into the main three categories of infectious, malignancies and auto-immune aetiology. Hemophagocytic lymphohistiocytosis (HLH) secondary to autoimmune causes is usually denoted as macrophage activation syndrome (MAS), an entity usually encountered by rheumatologists as a devastating condition complicating autoimmune diseases, most commonly systemic onset juvenile idiopathic arthritis (SJIA), adult onset still’s disease (AOSD) as well as lupus. The diagnosis of HLH is the first challenging critical step in its successful treatment due to its rarity, variable presentations and non-specific findings [3]. The diagnosis depends more on detecting a pattern of clinical and laboratory findings. A common mistake which delays diagnosis and hence treatment is looking to demonstrate hemophagocytosis, which may not be present at the onset, or in the site of biopsy, and which is neither sensitive nor specific for HLH. The pattern of clinical and laboratory finding are characteristic of hypercytokinemia, with fever being caused by the interleukins and tumor necrosis factor alpha (TNF-a), activated macrophages producing ferritin and hence hyperferritinemia, and plasminogen activators causing hyperfibrinolysis and thus hypofibrinogenemia. Lipoprotein lipase is suppressed causing hypertriglyceridemia and cytokines also suppress hemopoiesis [4]. What further complicates an already complicated issue is that spectrum of HLH manifestations can range from an atypical infectious mononucleosis like disease especially when caused by Epstein–Barr virus (EBV), the most frequent infection associated with HLH (EBV-HLH), to an abrupt devastating course, and that it can also manifest as organ specific illness like liver disease, coagulopathies like DIC, bone marrow failure, skin manifestations, pulmonary symptoms, neurologic symptoms, ophthalmic symptoms, and even as PUO [3]. This becomes problematic especially for the rheumatologist who is accustomed to seeing the abrupt devastating type of presentation more often. Guidelines for diagnosis of HLH were first formulated in 1991. On recognition that all the criteria may not be present in some patients, or may be dispersed over time, the diagnostic criteria were further revised in 2004 to include absent or low levels of natural killer cell activity, hyperferritinemia, and increased levels of soluble Interleukin 2 receptor (sIL2r) otherwise called named Cluster of Differentiation 25 (CD25). A diagnosis required five of the eight criteria to be fulfilled, but patients with molecular diagnosis of HLH did not need to fulfill the five criteria [5]. The relation of EBV with HLH is complex and yet exceptional. A ubiquitous virus, EBV affects more than 90% of the population. Primary infection is asymptomatic but exceptionally causes infectious mononucleosis (IM) in young adults and children. It stays latent in B cells and nasopharyngeal cells. Other associations are with tumors like nasopharyngeal carcinoma (NPC), Gastric carcinoma, Burkitt lymphoma, Hodgkin’s lymphoma and in immuno-deficient patients with B cell lymphoma. Rarer manifestations are EBV associated HLH (EBV-HLH) and chronic active EBV infection called CAEBV. These two manifestations are characterized by infection of cells other than B cells. In EBV-HLH, T cells, mostly CD8+ but also CD4+, and natural killer (NK) cells are the target of infection. Delayed infection of the B cells also occurs during the disease apart from itself being a reservoir [6]. Infection of T-lymphocytes, mostly CD8 cells by EBV upregulates the pro-inflammatory cytokines and plays a crucial part in HLH pathogenesis. The viral product responsible for the upregulation of cytokines, especially TNF-alpha is the LMP-1 protein. By suppressing the TNFR-1 and blocking the apoptotic signaling, it confers resistance to cytokine mediated injuries, and thus a sort of immortality, leading to progression of disease from HLH to T cell lymphoma [7]. The CD21 in the B cells is the receptor of C3d and also acts as the EBV receptor. In vitro, the B cell gets transformed and develops the ability to proliferate independently indefinitely. Even in the latent infections, viral antigens continue to be expressed. Memory B cells are the reservoir and even on treatment with acyclovir, the virus persists in the B cells. However, only a small fraction of B cells produces the virus while retaining the ability to produce antibodies. Four types of latencies have been described in EBV infection. Type 0 is the true latency characterized by persistence of EBV in memory cells. Type 1 in Burkitt and gastric lymphoma in which EBV cells migrate to daughter cells. Type 2 found in Hodgkin’s disease, natural killer T cells lymphoma and NPC in which protein required for transformation are lacking. Type 3 is called growth program because of its ability to induce autonomous proliferation of lymphoblastoid cell lines (LCLs) in vitro. The fourth is the CAEBV which is more common in the Asian population, found in immuno-competent hosts, and is characterized by persistence of EBV infection and high viral loads and unusual antibody pattern of elevated IgG VCA, negative IgM VCA and positive anti-EBNA [8]. For control of EBV infection, cellular immunity has a greater role. In the initial phase, suppressor T cells, NK cells and non-specific cytotoxic T cells are important; while HLA restricted cytotoxic T cells recognize the EBV associated large membrane proteins (LMP) and destroys these cells. If T cell immunity is compromised, EBV-B cells can start proliferating even in otherwise immunocompetent persons. This process is one of the many steps leading to neoplastic transformation. The LMP of EBV mimics tumor necrosis factor (TNF) a receptor and transmits growth signals which may be one of the mechanisms leading to the neoplastic transformation [9]. Hence in EBV-HLH treatment of T cell abnormalities without treatment of B cell- EBV infection can lead to recurrences and potentially to EBV induced lymphoproliferative diseases (LPD) later. In addition, T and NK cell LPD is found in the Asian subset of patients due to infections of these cells by EBV [10]. The EBV-HLH can range from spontaneously resolving inflammation to devastating and life-threatening disease [3]. Epstein–Barr virus (EBV) may also precipitate HLH in the patients with the genetic form of the disease. Skin manifestations of HLH are not well known specially to rheumatologists. They are highly pleomorphic and include generalized maculopapular erythematous rashes, generalized erythroderma, edema, panniculitis, morbilliform erythema, petechiae, and purpura with the incidence ranging from 6–65% in various series. The EBV–HLH has high mortality rate and optimal treatment is still not known. Many reports and present understanding describe good outcomes using immunosuppression to inhibit overactive T and NK cells and chemotherapy to target dividing lymphocytes. Reports also suggest that if the inciting pathogen (EBV) is targeted, the outcomes improve. In EBV-HLH, there is deficient clearing of EBV infected B cells in-vivo. It is thus hypothesized that by reducing the load of circulating EBV infected cells, the triggering of HLH can be prevented. In this context, anti-CD 20 therapy which targets B cells becomes a meaningful option in treatment of EBV-HLH, along with corticosteroids, CsA and etoposide which targets the T cell part of the pathology [10]. A retrospective study showed that adding rituximab improved the clinical status in 43% of the patients [11]. The treatment of acquired HLH in this study was based on 2004 HLH protocol with corticosteroids and cyclosporine along with anti–CD20 therapy to clear the infected B cells. In our patient, the skin manifestation of panniculitis was the only manifestation for which the patient had presented. She was however referred to the rheumatology clinic due to myositis on EMG testing. The panniculitis was probably thought to be due to rheumatological cause. Retrospectively it is difficult to opine whether the myositis too was caused by EBV primarily or by low grade EBV-HLH. Inflammatory myositis has been reported in HLH due to AOSD as reported by Umeda et al. [12] and myositis is rare in AOSD per se though reported by Monero-Alvarez et al. and Yanai et al. [13][14]. As EBV also causes myositis, the question remains open as to the cause of myositis in our patient; either EBV or EBV-HLH. There are few limitations in our case. Firstly, hemophagocytosis was not demonstrated in any tissue sample and though our case fulfilled 5 of 8 criteria required for the diagnosis, soluble CD 25, NK-T cell activity and genetic diagnosis were not done. Histologic evidence of hemophagocytosis is not specific for HLH and can be found in conditions like blood transfusion, sepsis, and chemotherapy. Also, only 58% of pediatric patients with final diagnosis of HLH were found to have histologic evidence of hemophagocytosis. The sensitivity and specificity of hemophagocytosis in diagnosis of HLH was 83% and 60% respectively. Moreover sCD25, NK-T Cell activity and genetic testing are done at specialized centers and are hence not helpful in acute settings requiring prompt decision making [15]. Recently, a diagnostic score called HS score was developed and validated to be used for estimating risk of having reactive hemophagocytic syndrome [16]. Retrospective application of the score to our case gave a score of 227 with 97.54% probability of the patient having hemophagocytic syndrome. Secondly, etoposide was not administered to our patient. Though it is a part of HLH 1994 and 2004 guidelines protocol the treatment of EBV-HLH is not formalized and involves treatment of the T cell as well as the B cell components including the trigger, i.e. the EBV infected B cell. Etoposide is T cell specific, and our case responded dramatically to treatment with methylprednisolone combined with cyclosporine and followed by dexamethasone. Cyclosporine too affects the T cell arm of the immunity and though etoposide was not used, the patient has had no recurrences and has remained asymptomatic even after two years of treatment with rituximab. Two pertinent questions can also be raised in our case. The first is regarding the simultaneous presence of both IgG as well as IgM VCA EBV antibodies which would certainly make it appear that the EBV infection was recent and occurred at the time the patient was diagnosed as HLH. However, the serological diagnosis of EBV is complex, can be difficult to interpret and many issues are yet unresolved. It is of importance to note that simultaneous presence of IgG as well as IgM VCA antibodies in the same patient can also be seen in reactivation in addition to recent infections [17]. This is in consonance with our view that the recurrent panniculitis in our patient was due to low grade HLH which resulted whenever EBV reactivation occurred. Whether EBV infection is acute or chronic can be known by testing for IgG avidity, identifying the class of antibodies by immunoblotting, looking for heterophile antibodies, anti-EA (D) antibodies or viral genome by molecular biology. As these tests were not carried out it can be seen to constitute a limitation in our case. However, these tests are not done at our center and getting it done from commercial laboratories would have greatly increased the cost to the patient. Another question which can be raised is whether treatment with rituximab was necessary in view of the good response to treatment by cyclosporine and steroids. It has been noted that some patients can have self-resolving HLH which can become recurrent later on to the extent of requiring immunochemotherapy and even hematopoietic cell transplantation. Ability of rituximab to eliminate EBV infected B cells makes it an useful addition to other treatments of EBV-HLH [3][4]. Moreover, as stated prior in the discussion, treatment of the B cell component in EBV-HLH can prevent recurrences and potentially EBV induced LPD later. We believe that treatment with rituximab was necessary in our case to eradicate the EBV and thus eliminate the antigenic stimulus causing HLH. This is also borne out in retrospect by the fact that the patient has had no recurrence of the panniculitis in over two years of follow-up after completion of anti–CD20 therapy. | ||||||
|
CONCLUSION
| ||||||
|
We conclude that hemophagocytic lymphohistiocytosis (HLH) may not always have a devastating presentation but can be low grade and have varied manifestations relating to different organ systems including panniculitis like in our case which the clinician has to be aware of. The evidence of EBV infection has to be actively sought in every patient of HLH, and if found should be treated with antiCD20 therapy in addition to the standard regimen. Our case illustrates the necessity of integrating anti–CD20 therapy into the treatment guidelines of HLH. | ||||||
|
REFERENCES
| ||||||
| ||||||
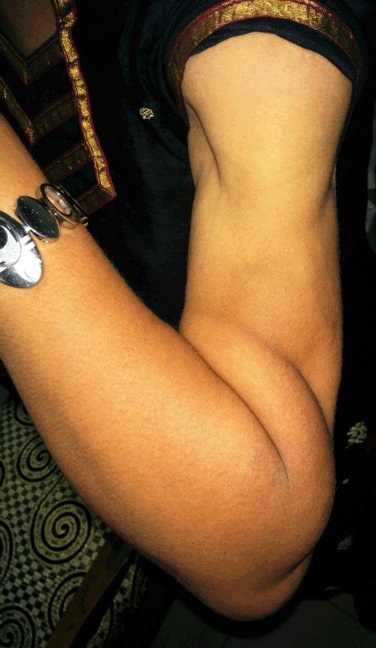

|
[Abstract HTML]
[Full Text HTML]
[Full Text PDF]
|
|
Author Contributions
Bijit Kumar Kundu – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Deepak Rath – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published |
|
Guarantor of Submission
The corresponding author is the guarantor of submission. |
|
Source of Support
None |
|
Conflict of Interest
Authors declare no conflict of interest. |
|
Copyright
© 2018 Bijit Kumar Kundu et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information. |
|
|