| |
|
|
|
Case Report
| ||||||
| An unusual case of a bicuspid aortic valve and non-compaction of the left ventricle | ||||||
| Abdullah Omar1, George Stoupakis2 | ||||||
|
1MD, PGY-1, Department of Medicine, Georgia Health Sciences University, Augusta, GA.
2MD, Interventional Cardiologist, Department of Medicine, Hackensack University Medical Center, Hackensack, NJ. | ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article]
[Similar article in Pumed] [Similar article in Google Scholar] 
|
| How to cite this article: |
| Omar A, Stoupakis G. An unusual case of a bicuspid aortic valve and non-compaction of the left ventricle. International Journal of Case Reports and Images 2013;4(5):279–282. |
|
Abstract
|
|
Introduction:
Myocardial noncompaction is thought to be a rare genetic disorder of myocardial morphogenesis.
Case Report: We report the case of a 22-year-old male with left ventricular noncompaction, bicuspid aortic valve and ascending aortic dilation. We discuss the genetic basis of myocardial noncompaction and potential etiologies that manifest in hyper-trabeculated myocardium in the presence or absence of extracardiac abnormalities. Conclusion: Myocardial noncompaction is a rare genetic disorder thought to arise from disruption of normal organogenesis during development. Many genetic abnormalities have been associated with myocardial non-compaction. Recent research prompts the question of whether various genetic mutations exert their effect via a common pathway which manifest in similar morphological findings. | |
|
Keywords:
Myocardial noncompaction, Bicuspid aortic valve, Myocardial morphogenesis, Genetics
| |
|
Introduction
|
|
Myocardial noncompaction (MNC) is a rare genetic disorder thought to result from an error in myocardial morphogenesis. During normal organogenesis, the meshwork of trabeculations on the luminal aspect of the heart compact to form the muscular walls. Myocardial noncompaction is thought to arise when there is an arrest of this compaction process. [1] Myocardial noncompaction is classified as nonisolated in the presence of extracardiac abnormalities and isolated in the absence of such abnormalities. [2] Noncompaction most commonly involves the left ventricle, referred to as left ventricular noncompaction (LVNC); however, it can also present in the right ventricle. The criteria used to define LVNC as proposed by Stöllberger et al. require more than three prominent trabeculations from the apex to the papillary muscles which have the same echogenicity as the myocardium and move synchronously with it; these trabeculations are not attached to the papillary muscles and are surrounded by intertrabecular recesses that perfuse directly from the ventricular cavity. [3] Alternatively, Petersen et al. considered the noncompacted to compacted (NC:C) ratio of myocardium on cardiac magnetic resonance imaging (MRI), and reported that a ratio of 1:2.3 in diastole accurately diagnosed LVNC. [4] Various genetic mutations have been implicated in the pathogenesis of noncompaction. According to a study of 34 patients with MNC, some of the major complications included heart failure (53%), ventricular tachycardia (VT) (41%) and thromboembolic events (24%). [1] |
|
Case Report
|
|
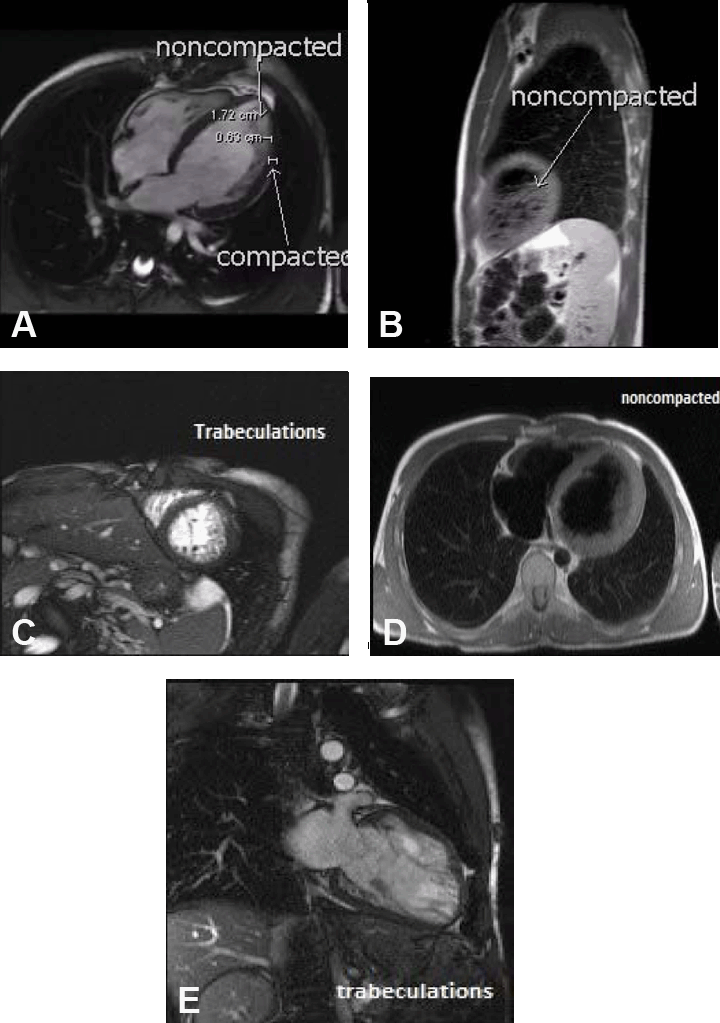
We report a case of a 22-year-old Caucasian male who presented to our emergency room (ER) with complaints of a sudden pounding of his chest, severe left shoulder pain, shortness of breath, dizziness and diaphoresis which started shortly after walking. Emergency medical services was called and on arrival found the patient in sustained VT at rate of 300 beats/min. In the field, he received three boluses of adenosine and one bolus of lidocaine without response and was subsequently cardioverted to normal sinus rhythm. Upon arrival to the ER, his vital signs were stable and he was started on aspirin, metoprolol, and intravenous amiodarone. He had no prior episodes of syncope or VT and his past medical history included asthma, premature birth and congenital, left conductive hearing loss. He smoked one pack per day (2–3 pack yrs) and had a maternal uncle with early myocardial infarction (age of male unknown). Physical examination revealed short stature and 2/6 diastolic murmur which was loudest at the left sternal border. He did not have hypermobile joints, arachnodactyly or any other cardinal features of Marfan's syndrome. Electrocardiogram (ECG) of the patient in the ER revealed normal sinus rhythm at 66 beats/min with a normal axis, left ventricular hypertrophy (LVH) and ST depression in V4–V6. His first troponin 1–2 hours after onset of symptoms was negative. The patient was urgently taken for cardiac catheterization and found to have normal coronary arteries. An aortogram showed a severely dilated ascending aorta with severe (4+) aortic regurgitation. A bedside echocardiogram showed LVH with preserved ejection fraction of 60% without wall motion abnormalities. He had a calcified bicuspid aortic valve (BAV) with mild stenosis and severe regurgitation. Aortic valve area by continuity equation measured 1.6 cm2, the mean gradient was 28 mmHg which was, consistent with mild aortic stenosis. In the left ventricle there were myocardial trabeculations with intertrabecular recesses suggestive of myocardial noncompaction. A cardiac magnetic resonance imaging (MRI) was done to evaluate the cardiac anatomy and demonstrated hypertrabeculations of the myocardium extending from the mid lateral wall and base towards the apex, with a ratio of NC:C myocardium exceeding 2.3:1. (Figure 1A-E) In addition, the ascending aorta at the level of right main pulmonary artery measured 4.6 cm and the calculated aortic regurgitant fraction was 50%. The patient was diagnosed with severe aortic valvular disease and noncompaction of the left ventricle. He was evaluated by cardiothoracic surgery and underwent an aortic valve replacement. |
|
|
|
Discussion
|
|
Isolated myocardial noncompaction is a rare genetic disorder with a reported prevalence of 0.05%; [5] however, the true prevalence is believed to be higher. During normal embryogenesis the mesoderm gives rise to a meshwork of loosely interwoven trabeculations in the luminal aspect of ventricular walls which condense from base to apex and from epidcardium to endocardium to form a compact wall. [6] The remaining intertrabecular recesses are reduced to capillaries during development. Left ventricular noncompaction is thought to arise from genetic abnormalities which lead to the arrest of this myocardial morphogenesis. Many studies have investigated different genetic mutations that present morphologically as noncompacted myocardium and other associated extracardiac defects. The notion that these different genetic mutations perhaps converge in a common pathway to manifest in a similar morphological profile has not been well studied. Various mutations have been found in patients with LVNC. Klaasen et al. studied several genes and suggested that isolated noncompaction lies within the diverse spectrum of cardiomyopathies triggered by gene defects in sarcomere proteins. [2] Our literature search revealed several reports linking chromosome 22q11 deletion with myocardial noncompaction, BAV and various aortic arch anomalies. Madan et al. reported a case of a young male with LVNC, BAV, and aortic dilation who was found to have a 22q11.2 deletion. [7] They also attributed the physical finding of bilateral conductive hearing loss to this deletion. Although our patient did not undergo chromosomal testing, we postulate his LVNC, BAV, dilated ascending aorta and conductive hearing loss (though unilateral) may have been the manifestation of a 22q11.2 deletion. In a study of mice, Kern et al. evaluated the role of the protease ADAMTS9 which plays an important role in cardiovascular development in mice. [8] During embryogenesis, ADAMTS9 was expressed in myocardial as well as extracardiac vascular smooth muscle cells. Later, expression of ADAMTS9 continued in the adult heart and the ascending aorta. They demonstrated that ADAMTS9 haploinsufficiency led to reduced cleavage of versican, an extracellular matrix glycoprotein. Some of the resulting phenotypic manifestations included hypertrabeculations as well as valvular and aortic wall anomalies. Similar cardiovascular abnormalities have been reported in humans with mutations in Notch genes. A highly conserved signaling pathway, Notch genes have been implicated in cardiomyocyte differentiation, valvular development and ventricular trabeculations. [9] Mutations in Notch genes have been associated with BAV. [10] Although the role of Notch in cardiac chamber abnormalities have not been fully elucidated, they are implicated in the endocardium-to-myocardium signaling which influences proliferation and differentiation of trabecular myocytes. Mutations in Notch have been linked with decreased trabecular myocyte proliferation via reduced BMP10 expression. The array of cardiac and extracardiac lesions resulting from singlegene defect begs the question of whether this concept could be the underlying cause of LVNC. Although the pathogenesis of myocardial hypertrabeculations is thought to be polygenic in nature, there may be a common signaling pathway defect like Notch or ADAMTS9 that interferes with trabecular compaction as well as with other cardiovascular and valvular processes as seen in non-isolated LVNC. |
|
Conclusion
|
|
Myocardial noncompaction is a rare genetic disorder thought to arise from an intrinsic defect in the process of morphogenesis. The defect is commonly attributed to a specific gene or gene cluster i.e. sarcomeric or structural genes. Perhaps the various mutations associated with MNC, whether in sarcomeric genes or structural genes, exert their effect via a common pathway that result in analogous cardiac and extracardiac anomalies regardless of the inciting mutation. Further research is warranted to answer many questions regarding the etiology and correct classification of myocardial noncompaction. |
|
References
|
|
|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions
Abdullah Omar – Substantial contributions to conception and design, Analysis and interpretation of data, Drafting the article, revising it critically for important intellectual content, Final approval of the version to be published Geroge Stoupakis – Substantial contributions to conception and design, Drafting the article, revising it critically for important intellectual content, Final approval of the version to be published |
|
Guarantor of submission
The corresponding author is the guarantor of submission. |
|
Source of support
None |
|
Conflict of interest
Authors declare no conflict of interest. |
|
Copyright
© Abdullah Omar et al. 2013; This article is distributed the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any means provided the original authors and original publisher are properly credited. (Please see Copyright Policy for more information.) |
|
|