|
|
|
|
Case Report
| ||||||
| Congenital nephrogenic diabetes insipidus presenting after acute pyelonephritis | ||||||
| Christian Castillo1, Poonam Bherwani2, Evelyn Erickson3, Gerard Prosper4 | ||||||
|
1Neonatology Fellow, Cook County Hospital, Chicago, Illinois, USA.
2Department of Pediatrics, Lincoln Medical and Mental Health Center, Bronx, New York, USA. 3Research Assistant, Department of Pediatrics, Lincoln Medical and Mental Health Center, Bronx, New York, USA. 4Attending of Pediatric Nephrology, Lincoln Medical and Mental Health Center, Bronx, New York, USA. | ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article]
[Similar article in Pumed] [Similar article in Google Scholar] 
|
| How to cite this article: |
| Castillo C, Bherwani P, Erickson E, Prosper G. Congenital nephrogenic diabetes insipidus presenting after acute pyelonephritis. International Journal of Case Reports and Images 2012;3(11):25–27. |
|
Abstract
|
|
Introduction:
Diabetes insipidus (DI) is characterized by the inability to concentrate urine. While central DI is caused by failure to release enough functional vasopressin, nephrogenic DI (NDI) is due to the insensitivity of the distal nephron to the effect of antidiuretic hormone (ADH).
Case Report: A 5-day-old newborn male was admitted for isolated fever and a questionable early right upper lobe infiltrate. He gradually developed hypernatremia and increased osmolality. As part of his work up for fever, he had a urine culture of 30K colonies of Enterococcus faecalis. His vasopressin test was negative. Conclusion: The polyuria and polydipsia associated with genetic NDI usually presents within the first several weeks of life but may only become apparent after weaning or with longer periods of nighttime fasting. The acute pyelonephritis of this newborn may have been the initial trigger for the congenital NDI. Accurate diagnosis of this patient helped to also diagnose his maternal uncle and provide clues to the current condition of his maternal grandmother. Early diagnosis and management can prevent the development of neurological and developmental complications associated with NDI. | |
|
Key Words:
Congenital Diabetes Insipidus, Nephrogenic Diabetes Insipidus, Acute Pyelonephritis
| |
|
Introduction
| ||||||
|
Diabetes insipidus is characterized by the inability to concentrate urine. While central DI is caused by the failure to release enough functional vasopressin (antidiuretic hormone), NDI is due to the insensitivity of the distal nephron to the antidiuretic effect of ADH. If presented at birth, it is considered a genetic mutation called congenital nephrogenic DI. [1] We present here a case of a newborn admitted for acute pyelonephritis with a normal chemistry, who subsequently developed hypernatremia and hyperosmolality. Acute pyelonephritis, being an interstitial nephritis may present with a renal tubular defect affecting the different segments. One of the manifestations is the loss of concentrating ability which is transient and resolves in a few weeks. | ||||||
|
Case Report
| ||||||
|
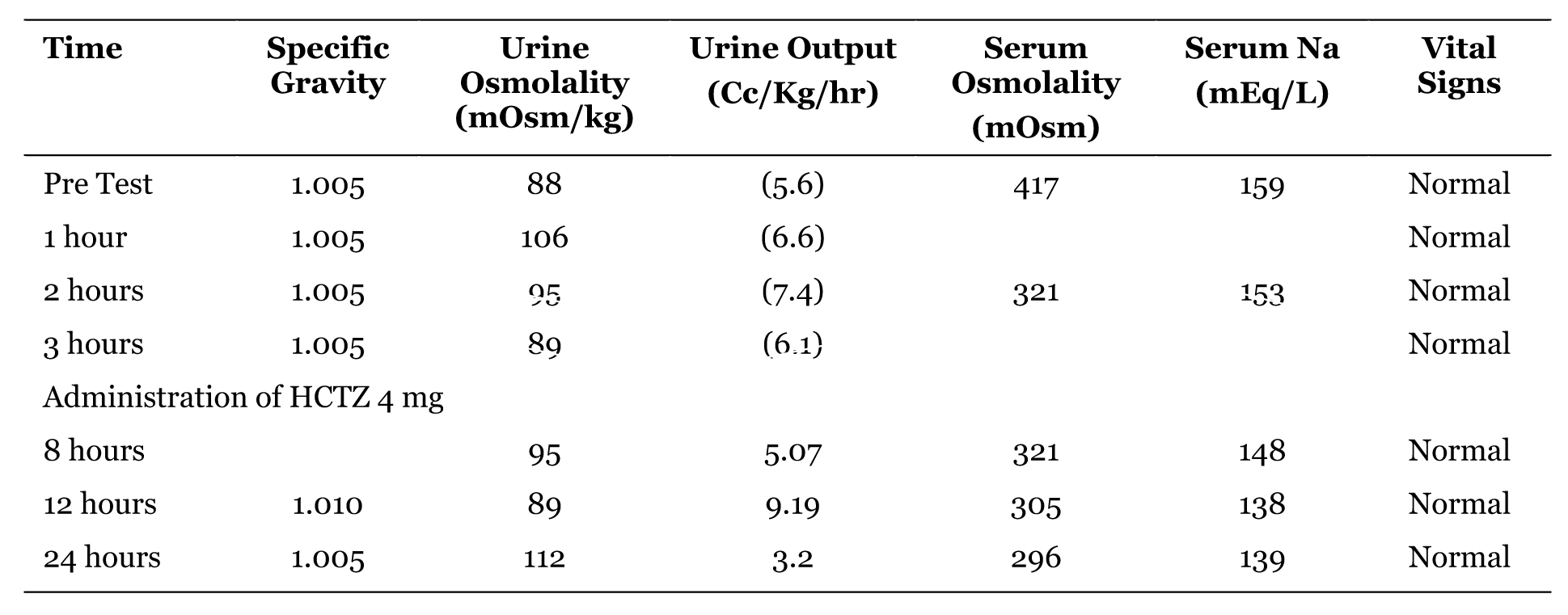
A 5-day-old newborn male, product of a full term and uncomplicated pregnancy was admitted to the hospital for fever without focus. All maternal ancillary tests including Group B Streptococcus were negative. On admission, the patient was well hydrated. His vital signs were temperature of 102.8 °F, pulse of 162, blood pressure of 86/47 mmHg and respiratory rate of 51, and O2 saturation of 95%. The physical examination was unremarkable. A complete sepsis work up was performed. CBC and CSF chemistry, and hematology as well as Blood Urea Nitrogen, and serum creatinine (BUN/Creat) were normal. However, the serum sodium was in the upper limit of normal and the urine specific gravity was less than 1.005. The patient was on formula feedings supplemented with IV Dextrose 5%/NaCl 0.2 (2/3 maintenance) and started on IV cefotaxime and ampicillin. On the 3rd hospitalization day, it was noted that the patient urine output was greater than 6 cc/kg/hr and the patient serum sodium went up to 159 mEq/L but BUN/Creat were normal. The serum osmolality increased to 417 mOsm and the corresponding urine osmolality was 88 mOsm/kg. The urine culture from a catheterized specimen grew 30K colonies of Enterococcus faecalis, which was sensitive to ampicillin. His fluid requirement was changed to IV Dextrose 5%/NaCl 0.2 (free water loss + daily maintenance) and oral feeding. To confirm the concentrating defect and to distinguish the renal form from the central form, a vasopressin test was performed. The test consists of the administration IV of 1 Unit of AVP by m2. The urine specific gravity, urine osmolality and output, serum osmolality, and serum sodium were determined pre and post administration of AVP and follow up monitoring at 1, 2 and 3 hours (Table 1). The results suggested the diagnosis of NDI which was later confirmed by an elevated level of AVP (15.2 pg/mL). | ||||||
| ||||||
|
Discussion
| ||||||
|
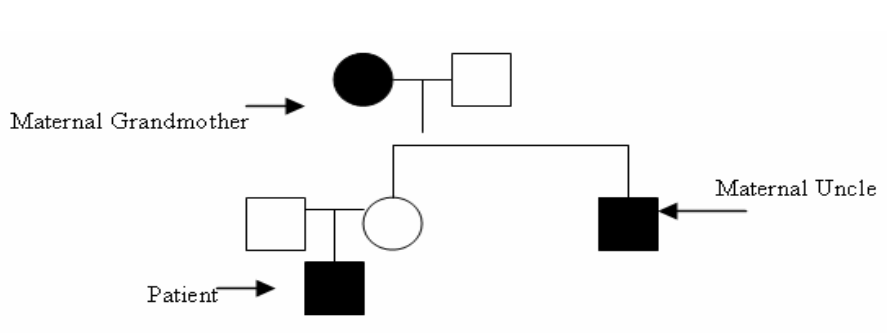
Three different inheritance patterns have been recognized. In most cases, approximately 90% are transmitted as an X-link recessive trait in the families. Female carriers were clinically unaffected, transmitting the disease to their sons who display the complete clinical picture. In 1998, the major NDI locus was mapped in the distal region of the long arm of the X chromosome (Xq28) and in 1992, the gene in VR2 was shown to underline X-linked NDI. [2] [3] In a minority of families, approximately 10% of the transmission and phenotypic characteristics of NDI are not compatible with an X-linked trait. In these families, females display a complete clinical picture of NDI and are clinically indistinguishable from affected males. In addition, linkage analysis in these families has excluded linkage between NDI and polymorphic DNA marker from the Xq28 region, a family pedigree has suggested the existence of both autosomal recessive and dominant forms of NDI. In the recent years, it has been demonstrated that both autosomal forms are caused by a mutation of an AVP sensitive AQP2 water channel. [4] In our patient, the genetic analysis reveals an AVPR2 sequence variant on the chromosome locus associated NDI. His maternal uncle was found to have the same mutation (Figure 1). For the maternal grandmother who was not tested but has clinical evidence of NDI, the most likely explanation for the existence of the phenotype carrier of the AVPR2 mutation varying from no symptom to complete manifestation of the disorder could be attributed to a skewed X inactivation. [5] The polyuria and polydipsia associated with genetic NDI usually presents within the first several weeks of life but may only become apparent after weaning or with longer periods of nighttime fasting. Many infants initially present with fever, vomiting, dehydration and failure to thrive. Acquired NDI may result from hypercalcemia or hypokalemia, and is associated with the following drugs: lithium, demeclocycline, foscarnet, clozapine, amphotericin, methicillin, and rifampin. In our case, the acute pyelonephritis may have been the initial trigger for the congenital NDI. [1] Initially, it was assumed that this case of NDI could have been caused by an acute pyelonephritis which is associated with tubular dysfunction as a disturbance of water and electrolytes imbalance. On further investigation, the family medical history revealed that the patient's maternal uncle who is 9 years old, has a history of polyuria and polydipsia. In addition, the maternal grandmother has a similar history since childhood, and reports that she drinks about 4 to 5 gallons of water a day. A genetics test coding regions of the AVP gene and the AVP receptor 2 (AVPR2) genes were sequenced in the outpatient setting revealing an AVPR2 sequence variant on the X chromosome associated with NDI. The maternal uncle was also tested and found to have similar results. | ||||||
| ||||||
|
| ||||||
|
Conclusion
| ||||||
|
Disturbance of water and electrolyte balances in acutely sick newborn and infant deserve an urgent systematic and detailed investigation for accurate diagnosis and proper management in view of preventing the neurological and developmental complications that could occur. Our patient had early manifestation of congenital NDI, contrary to his uncle who developed symptoms at a later age and the grandmother who appears to have a mild form. Acute pyelonephritis may be a compounding factor for the initiation of his NDI. For it is known that acute pyelonephritis may cause an acquired and transient NDI by downregulating AQP2 expression. Accurate diagnosis of this patient helped to diagnose his maternal uncle with hereditary NDI due to an AVPR2 x-linked mutation, and provide clues to the maternal grandmother's clinical manifestation. | ||||||
|
References
| ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions:
Christian Castillo - Conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Critical revision of the article, Final approval of the version to be published Poonam Bherwani - Conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Critical revision of the article, Final approval of the version to be published Evelyn Erickson - Acquisition of data, Drafting the article, Critical revision of the article, Final approval of the version to be published Gerard Prosper - Conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Critical revision of the article, Final approval of the version to be published |
|
Guarantor of submission:
The corresponding author is the guarantor of submission. |
|
Source of support:
None |
|
Conflict of interest:
Authors declare no conflict of interest. |
|
Copyright:
© Christian Castillo et al. 2012; This article is distributed the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any means provided the original authors and original publisher are properly credited. (Please see Copyright Policy for more information.) |
|
|