
|
 |
|
Case Report
| ||||||
| Churg–Strauss syndrome (eosinophilic granulomatosis with polyangiitis): A case report | ||||||
| M. West1, P. Kumar1 | ||||||
|
1Department of Medicine, Mackay Base Hospital, Australia
| ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article] [Similar article in Pumed] [Similar article in Google Scholar] 
|


| How to cite this article |
| West M, Kumar P. Churg–Strauss syndrome (eosinophilic granulomatosis with polyangiitis): A case report. Int J Case Rep Images 2017;8(8):555–560. |
|
ABSTRACT
| ||||||
|
Churg–Strauss syndrome (CSS) is a rare systemic vasculitis of the small and medium sized blood vessels. The triad of asthma, sinusitis and hypereosinophilia is characteristic of CSS. However, it can affect any organ system with predominance for the skin, respiratory, neurological, gastrointestinal and cardiovascular systems. The natural history of the condition has been described in three phases: prodromal, eosinophilic and vasculitic. Depending on the organ system affected and stage of the disease, the presentation of CSS can be varied and the diagnosis can be challenging. The most frequently used criteria for diagnosing CSS are those developed by the American College of Rheumatology in 1990. Such classification criteria can assist in making the diagnosis of CSS and differentiating the condition from other diseases that cause pulmonary infiltrates with eosinophilia including allergic bronchopulmonary aspergillosis, acute and chronic eosinophilic pneumonia, idiopathic hypereosinophilic syndrome, certain parasitic infections and drug reactions. We present a case that is characterized by hypereosinophilia, vasculitis involving mainly the pulmonary and nervous systems, with a history of allergic rhinitis and sinusitis. Early diagnosis is crucial so that systemic glucocorticoids (the mainstay of treatment) can be commenced and can help to prevent organ damage and mortality. Keywords: Churg–Strauss syndrome, Eosinophilic granulomatosis polyangiitis, Pulmonary infiltrates | ||||||
|
INTRODUCTION
| ||||||
|
Eosinophilic granulomatosis with polyangiitis (EGPA), or more commonly known as Churg-Strauss syndrome (CSS) is a necrotizing small and medium vessel vasculitis associated with blood and tissue hypereosinophilia and usually occurring in people with a history of asthma [1]. Churg–Strauss syndrome is a rare disease with the annual incidence ranging from 2.4–6.8/1 million in the general population. It has equal gender distribution, average affected age ranges from 40–60 years old and there is no ethnic or familial predisposition [2]. Pathologists Dr Jacob Churg and Dr Lotte Strauss first described this condition as a distinctive clinical entity in 1951 [3][4]. Based on their pathological examinations and postmortem studies, they noted a syndrome of asthma, hypereosinophilia, necrotizing vasculitis and extravascular granulomas [3]. They identified it as related to, but distinct from Polyarteritis Nodosa (PAN), which they termed allergic granulomatosis and angiitis [3][5][6]. The pathogenesis of CSS is still unknown [2][7]. It is believed to be allergic or immune mediated due to the presence of allergic symptoms, elevated IgE levels and immune complex mediation [7][8] with 40–60% being antineutrophil cytoplasmic antibody (ANCA) positive (predominantly perinuclear ANCA with anti-myeloperoxidase specificity) [1]. Herein, we report on a case that fits the clinical criteria for diagnosis of Churg–Strauss syndrome, supported by classic laboratory findings (not always present) and imaging results. | ||||||
|
CASE REPORT
| ||||||
|
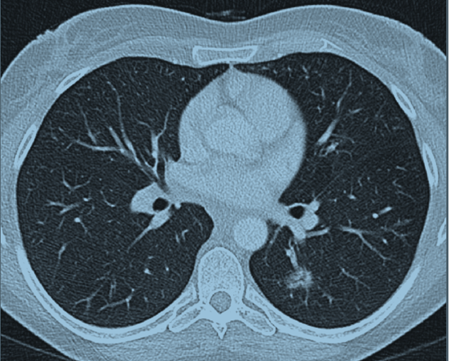
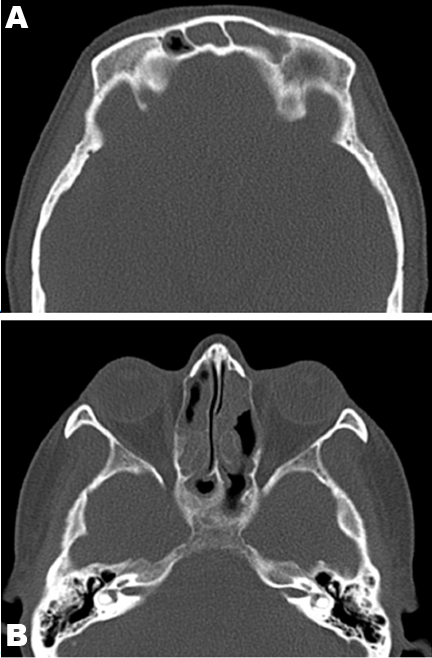
A 52-year-old female was referred by her general practitioner to the respiratory outpatients clinic (Mackay Base Hospital, Australia) for the evaluation of progressive dyspnea and wheeze. She reported a three-month history of increasing shortness of breath, with persistent wheeze, chest tightness and a dry cough. She also reported paroxysmal nocturnal dyspnea, but denied orthopnea or peripheral pitting edema. The patient had been treated with bronchodilators and antibiotics with minimal relief. On review of systems, the patient denied skin rashes, joint swelling, myalgias, arthralgias, sore throat, dysphagia, odynophagia, abdominal pain, and changes in bowel habits or urinary symptoms. She did report tingling, numbness and pain in her left arm. She also endorsed some weight loss of five kilograms over the last three months. However, there was no history of fever or night sweats. The patient’s past medical history was significant for allergic rhinitis and sinusitis for the last two years. There was no history of asthma. She was also recently diagnosed with Grade 2 cervical intramedullary astrocytoma. The tumor was not resectable, and so she was treated with radiotherapy and chemotherapy (temozolomide) with neurosurgical follow-up. She has also had a splenectomy in the past for idiopathic thrombocytopenic purpura. She suffered depression as well. There was no family history of asthma or atopy. Her mother had cervical cancer and her brother had hypertension. The patient had previously worked as a nurse. There was no exposure to organic or mineral dust, fumes or pets. There was no history of travel. She was a lifelong non-smoker, and rarely consumed alcohol. On examination, the patient had a pulse rate of 72 beats/min, blood pressure of 132/84 mmHg, saturations of 98% on room air and she was afebrile. On auscultation of the chest, there were vesicular breath sounds with a few bibasal crepitations and bilateral rhonchi throughout the lungs. Her heart sounds were dual with no murmurs. Her abdomen was soft and non-tender. Her calves were soft, non-tender and there was no pitting edema. No skin rashes were noted. Initial blood tests (reference ranges in defined opacities in the left lower lobe parentheses) of the patient showed hemoglobin level of 12.0 (11.5–16.0 g/dl), a white blood cell count of 10.3 cm3 (4–11 cm3) with a raised eosinophil count of 3.3 (<0.60). Electrolytes, urea, creatinine, liver function tests and coagulation profile of the patient were within normal ranges. The C-reactive protein (CRP) was 11 (<5), and erythrocyte sedimentation rate (ESR) was 21 (<19). Urine examination was normal. The patient’s routine chest X-ray was normal. Computed tomography (CT) scan of the chest revealed a focal ill-defined alveolar opacity in the peribronchial region of the left lower lobe apical segment and another smaller similar opacity in the left upper lobe centrally (Figure 1). Computed tomography scan of the sinuses (Figure 2) showed near complete opacification of the frontal and the visualized ethmoid sinuses with mild mucosal thickening throughout the maxillary, sphenoid, and ethmoid sinuses and hypertrophy of the turbinates. Spirometry showed a moderate obstructive pattern with forced expiratory volume (FEV1) of 58% predicted, forced vital capacity (FVC) of 74% predicted, and FEV1/FVC of 79%. There was severe small airway obstruction with maximum midexpiratory flow (MMEF 75/25) of 30% predicted. There was gas trapping with residual volume (RV) of 142%. Gas diffusion was normal with diffusing capacity (DLCO) being 89% and transfer coefficient of carbon monoxide (KCO) being 85%. A transthoracic echocardiogram showed normal left ventricular systolic function, ejection fraction of 65% and normal diastolic function. No valvular abnormalities. Subsequent bloods showed serum IgE was markedly elevated at 1140. Serum precipitins against Aspergillus fumigatus were negative. Bloods were positive for perinuclear antineutrophil cytoplasmic antibodies (p-ANCA) at a level of 640 by indirect immunofluorescence. The ANCA subsets directed towards the proteins myeloperoxidase (MPO-ANCA) were also positive at 417. Bloods were negative for cytoplasmic antineutrophil cytoplasmic antibodies (c-ANCA) and also negative for the subset directed towards proteinase-3 (PR3-ANCA). A bronchoscopy and bronchoalveolar lavage was done which showed normal respiratory flora and a cell count less than 10. No open lung biopsy was performed, as there were sufficient criteria for a clinical diagnosis of eosinophilic granulomatosis with polyangiitis or Churg–Strauss syndrome to be made. The patient was commenced on prednisolone (50 mg/day) and this was associated with a marked improvement in her symptoms. She reported no dyspnea or wheeze, and the neuropathic pain and paresthesia in her left arm decreased substantially. The prednisolone dose was slowly tapered and is still ongoing. Her last computed tomography scan of the chest (performed after three months of treatment with corticosteroids) shows complete resolution of the previously visualized areas of ground glass opacities in the left lower lobe. | ||||||
| ||||||
| ||||||
|
DISCUSSION
| ||||||
|
The triad of asthma, sinusitis and hypereosinophilia is suggestive of Churg–Strauss syndrome [4]. Churg–Strauss syndrome however can be highly variable in its presentation and course and depending on the organ systems involved [6]. The natural history of the disease has been described in three phases [1]:
As a systemic vasculitis, CSS can involve any organ system but predominantly involves skin, [1] respiratory, neurological, gastrointestinal and cardiovascular systems [4]. Poor prognosis is usually associated with gastrointestinal and cardiovascular involvement [1]. Common manifestations related to involvement of different organ systems are outlined below:
This case of CSS was characterized by hypereosinophilia, vasculitis involving mainly the pulmonary and nervous systems and with a history of allergic rhinitis and sinusitis. Interestingly, there was no history of asthma, which is considered the hallmark of CSS [12]. The diagnosis of CSS can be difficult especially given the variable presentation, and depending on the stage of CSS [1][2]. The laboratory findings are non-specific and include eosinophilia, high IgE and raised acute phase reactants. The ANCA positivity (particularly perinuclear ANCA of anti-myeloperoxidase specificity) is found in approximately 40–60% of patients [1]. Again, this is not specific to CSS, and can be associated with microscopic polyangiitis (MPA), another one of the vasculitides [8]. In this case, the patient had peripheral eosinophilia (3.3), markedly elevated serum IgE (1140), and p-ANCA positive (640), specifically MPO-ANCA positive (417). Although these findings are not specific, it is useful in supporting the diagnosis in conjunction with the other clinical findings. For clinical diagnosis of CSS, the diagnostic criteria published by the American College of Rheumatology (ACR) in 1990 are the most widely used [1][11]. The criteria are: asthma, eosinophilia (greater than 10% on white blood cell differential count), paranasal sinusitis, pulmonary infiltrates (non-fixed), extravascular eosinophils on biopsy, and neuropathy (mononeuropathy including mononeuritis multiplex or polyneuropathy) [11]. At least four out of the six criteria are required for a confident diagnosis of CSS [1]. Our patient satisfied four of the six criteria with peripheral eosinophilia, paranasal sinusitis, mononeuropathy (with neuropathic pain, numbness and paresthesia in left arm), and pulmonary infiltrates. This makes the diagnosis of CSS with 85% sensitivity and 99.7% specificity [11]. The other classification systems include Lanham’s criteria, which proposes a clinical diagnosis defined by the coexistence of asthma, blood eosinophilia (>1500 eosinophils/µl) and evidence of vasculitis in two or more extrapulmonary organs [2][13]. However, as this involves at least two sites of vasculitic involvement, it can miss patients early in the course of the disease and delay treatment [2]. The Chapel Hill Consensus conference also established a new classification system, which defines CSS as: ‘eosinophil-rich and granulomatous inflammation often involving the respiratory tract, and necrotizing vasculitis affecting small to medium-sized vessels associated with asthma and eosinophilia [14]. However, this definition has also been criticized as requires biopsy to establish diagnosis and lacks specificity in comparison to other classification systems [2]. The biopsy features proposed originally by Churg and Strauss [3] required the presence of:
However, this triad hardly ever coexists at the same time in any one CSS patient, which can delay diagnosis [2]. Although easily accessible tissues should be biopsied, the clinical features of CSS are so distinctive that tissue biopsy is invariably not required [15]. In this case, the diagnosis had been made according to the clinical criteria published by the ACR and so biopsy was not required. Churg–Strauss syndrome must be differentiated from other diseases that cause pulmonary infiltrates with eosinophilia including allergic bronchopulmonary aspergillosis, acute and chronic eosinophilic pneumonia, idiopathic hypereosinophilic syndrome, certain parasitic infections and drug reactions.
Early diagnosis and treatment is important for prevention of organ damage and mortality [1]. Systemic glucocorticoids are the mainstay of treatment [2]. However in a subset of patients, combined therapy with a cytotoxic agent (i.e. cyclophosphamide, azathioprine) may be required to control the disease [8]. Biologic agents such as rituximab, interferon-alpha, infliximab and etanercept are the focus of clinical trials for their potential role in CSS treatment in the future [2]. The prognosis of CSS is quite good in comparison to other systemic vasculitides, with a survival rate of 70% at five years [1]. | ||||||
|
CONCLUSION
| ||||||
|
In this case, the patient was commenced on high dose prednisolone with a subsequent improvement in symptoms, blood eosinophil count, inflammatory markers and resolution of pulmonary infiltrates on imaging. We will continue to slowly wean her prednisolone dose, with the possibility of commencing a steroid-sparing agent in the future if required. | ||||||
|
REFERENCES
| ||||||
| ||||||

|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions
M. West – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published P. Kumar – Analysis and interpretation of data, Revising it critically for important intellectual content, Final approval of the version to be published |
|
Guarantor
The corresponding author is the guarantor of submission. |
|
Source of support
None |
|
Conflict of interest
Authors declare no conflict of interest. |
|
Copyright
© 2017 M. West et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information. |
|
|