|
|
|
|
Case Report
| ||||||
| Familial congenital hypothyroidism due to thyroid dysgenesis: A case report of largest family | ||||||
| Abhinav Kumar Gupta1, Syed Mohd. Razi2, Deepak Chand Gupta1, Saqib Ahmad Khan1, Pankaj Jain3, Keshav Kumar Gupta4 | ||||||
|
1Senior resident, Department of Endocrinology, L.L.R.M. Medical College, Meerut, Uttar Pradesh, India 2Consultant endocrinologist, Department of Endocrinology, Sri Sai hospital, Muradabad, Uttar Pradesh, India 3Assistant Professor, Department of pediatrics, S.M.S. Medical College, Jaipur, Rajsthan, India 4Professor, Department of Endocrinology, L.L.R.M. Medical College, Meerut, Uttar Pradesh, India | ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article] [Similar article in Pumed] [Similar article in Google Scholar] 
|
| How to cite this article |
| Gupta AK, Mohd. Razi S, Gupta DC, Khan SA, Jain P, Gupta KK. Familial congenital hypothyroidism due to thyroid dysgenesis: A case report of largest family. Int J Case Rep Images 2017;8(8):549–554. |
|
ABSTRACT
| ||||||
|
Introduction:
Congenial hypothyroidism due to thyroid dysgenesis is usually regarded as sporadic. However, a small but significant proportion of familial cases have been identified (2%). Herein, we describe a case report of unusually large family of 10 siblings, out of which five were affected with congenital hypothyroidism, which is supposed to be the world’s largest series of familial congenital hypothyroidism due to thyroid dysgenesis. Keywords: Athyreosis, Familial congenital hypothyroidism, Sibling, Thyroid dysgenesis | ||||||
|
INTRODUCTION
| ||||||
|
Congenital hypothyroidism (CH) is one of the most common preventable causes of mental retardation. Incidence of CH was initially reported to be in the range of 1:3000 to 1:4000. With the help of national screening programs, it has become apparent that the incidence varies by geographic location. A recent report showed that the incidence in the United States increased from 1:4094 in 1987 to 1:2373 in 2002. Incidence varies among different racial and ethnic groups, gender, birth weight, single versus multiple births and according to mother’s age [1]. Eighty-five percent of cases of CH are caused by thyroid developmental defects (dysgenesis) and remaining 15% of cases occur due to the defects in thyroid hormone biosynthesis (dyshormonogenesis). The pathogenesis of thyroid dysgenesis (TD) is still unknown, and the disease is usually regarded as sporadic with a female predominance. Possible roles of autoimmune or unidentified environmental factors have been suggested but not confirmed. Some familial cases of CH caused by thyroid dysgenesis have been reported with either athyreosis or ectopic gland in affected members. French study reported a significant important proportion of familial cases of CH due to thyroid dysgenesis (2%), affirming the existence of strong familial component in CH due to thyroid dysgenesis [2]. | ||||||
|
CASE REPORT
| ||||||
|
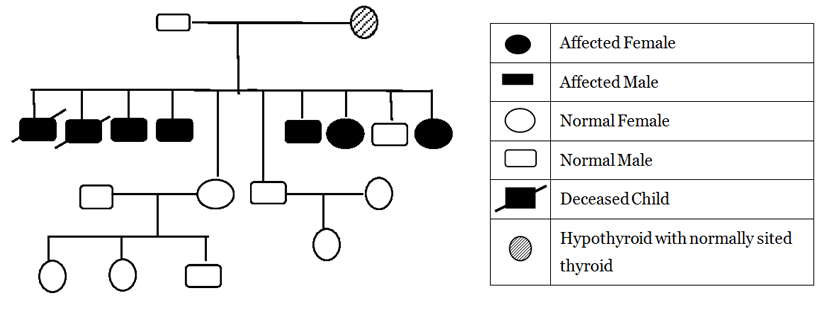
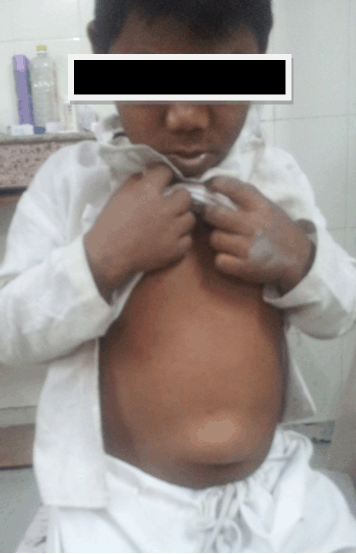
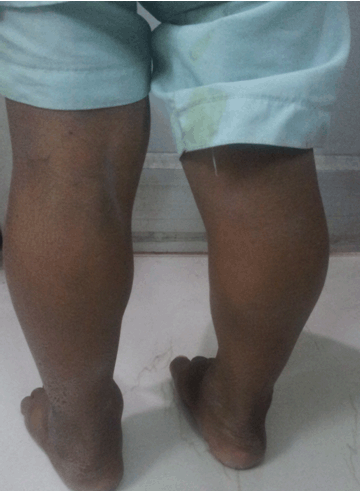
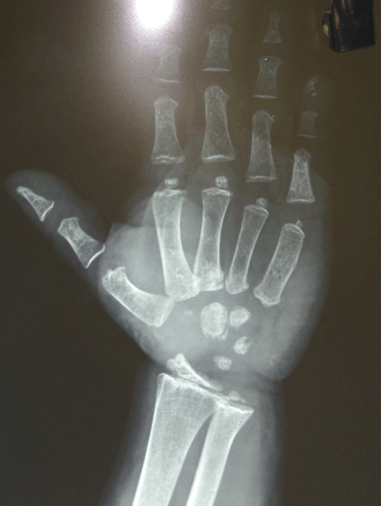
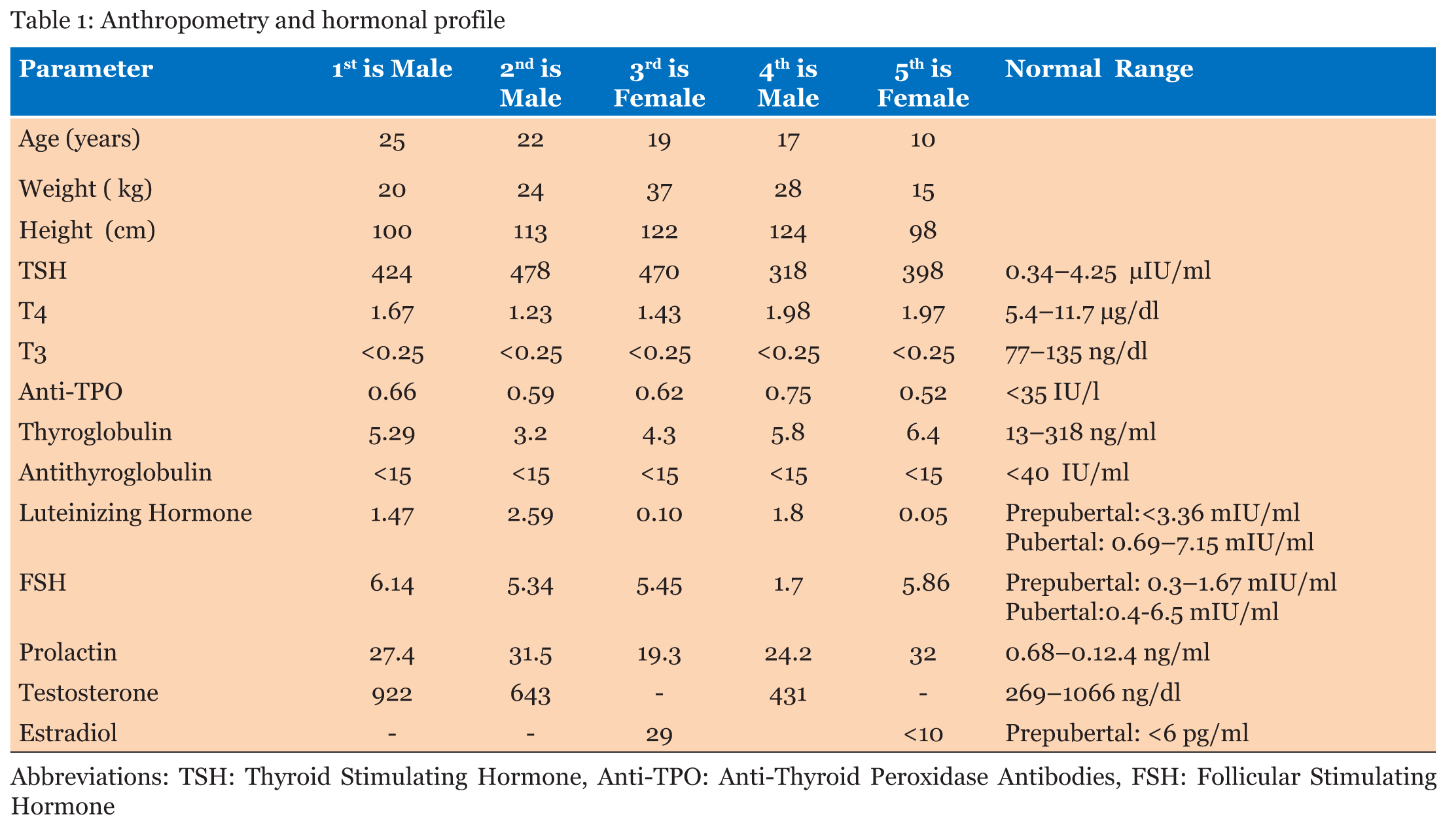
In this case report, we describe an Asian–Indian family of 10 siblings out of which five presented to endocrinology outpatient clinic with complaints of lethargy, constipation, hoarseness of voice, edema, short stature and mental retardation. The eldest affected sibling was 25-year-old while the youngest affected child was 10-year-old. All the children were born to a hypothyroid mother, out of a non-consanguineous marriage with full term normal vaginal delivery. The mother did not give history of thyroxin intake during any of the pregnancies. All the pregnancies and ante-partum periods were uneventful. There was no history suggestive of delayed cry, birth hypoxia, prolonged jaundice, in any of the affected siblings. All the affected siblings had feeding difficulties, delayed fine motor, gross motor and social developmental milestones along with subnormal intelligence. Detailed family history revealed demise of two siblings at the ages of two years and 32 years respectively other than these five affected siblings, with the similar complaints while the remaining three are normal (Figure 1). On general examination all the affected siblings were having typical puffy hypothyroid facies with cold dry skin, sparse fragile hair, lateral madarosis, short round nose, macroglossia, edema, hoarseness of voice and bradycardia (Figure 2). Calf muscle pseudo-hypertrophy and umbilical hernia were present in two of the affected siblings (Figure 3) and (Figure 4). There was absence of goiter, hearing difficulty, cleft palate, spiky hair, low posterior hairline, low set ear and abnormal movement. On anthropometric examination all affected siblings had significant short stature. Sexual maturity was corresponding with the chronological age in all affected siblings. Systemic examination of all patients was unremarkable except for delayed relaxation of deep tendon reflexes and pseudomyotonia. Laboratory investigations revealed normal hemogram, renal function test, hepatic function test, electrolyte, LH, FSH and prolactin. Tests also revealed very high level of TSH along with very low levels of T4 and T3. Anti-thyroid peroxidase antibody (Anti-TPO) and anti-thyroglobulin (Anti-TG) were negative in all siblings. Serum thyroglobulin was very low in all the affected siblings (Table 1). X-ray of left hand revealed significant delay in bone age and epiphyseal dysplasia (Figure 5). Ultrasonography of the thyroid gland revealed complete absence of thyroid tissue in all affected siblings. Maternal laboratory testing revealed normal anti-TPO and normally located thyroid gland on ultrasonography. Father’s thyroid function tests were normal. On Tc99m pertechnetate scan, there was no radioactive tracer uptake at any location. All the hormonal investigations were done by chemiluminescence immunoassay using Abbott ARCHITECT i1000sr immunoassay analyzer (USA). On the basis of typical clinical history, physical examination and investigations all five siblings were diagnosed as cases of CH. They were started on thyroxine replacement and dose titrated by periodic laboratory investigation. | ||||||
| ||||||
| ||||||
| ||||||
| ||||||
| ||||||
| ||||||
|
DISCUSSION
| ||||||
|
Congenital hypothyroidism is defined as reduced level of thyroid hormones, which may be symptomatic at birth or later. It may be either due to thyroid dysgenesis (abnormal gland development) or dyshormonogenesis (disorder of thyroid hormone biosynthesis). Congenital hypothyroidism is classified as permanent and transient. Permanent CH refers to a persistent deficiency of thyroid hormone requiring life-long replacement. Transient CH is deficiency of thyroid hormone at birth which recovers in neonatal period or later. Causes of transient CH include iodine deficiency, transfer of maternal blocking antibodies, fetal exposure to anti-thyroid drugs, maternal iodine exposure, neonatal iodine exposure and mutation in DUOX2 and DUOXA2 [1]. Most of the cases of permanent CH are related to TD due to abnormal thyroid gland development involving various defects, such as thyroid ectopy, athyreosis and thyroid hypoplasia. About 5% of the cases have been shown to result from the mutations in genes involved in thyroid development, but most cases of TD are sporadic and their pathogenesis remains unknown. A high frequency of congenital cardiac defect in TD support the role of genetic component and reports of some familial cases of CH due to TD resulting from athyreosis and ectopic gland is consistent with the notion of an inherited disease [3]. Anger and Kelley described unusual occurrence of athyreotic cretinism in three siblings of Maxican-American descent who were born out of consanguineous marriage [4]. Cross et al. observed two sisters with cretinism and Kocher–Debre–Semelaigne syndrome (myotonia and muscular pseudohypertrophy). Although no thyroid tissue was palpable but on investigation small thyroid tissue was present [5]. Kaplan et al. described two non-consanguineous Ashkenazi Jewish families, in each of which, a brother and sister had hypothyroidism, associated with ectopia and hypoplasia of the thyroid [6]. Yano et al. described athyreotic CH in two sisters born out of non-consanguineous parents in Japan [7]. Positive family history was found in 2% of CH patients with TD [8]. Castanet et al. compared familial and sporadic cases of CH. He identified 67 patients were having family history of CH with TD, out of them only two families had three or four affected members, all other families were having =2 affected members. He observed significantly higher number of familial cases than it would occur by chance only, hence drawn the conclusion that genetic factors could be involved in TD [2]. Almost 20 families of CH due to TD have been described in last 40 years, which again implies that TD may be familial. A number of genes which are expressed during thyroid embryogenesis have been implicated in TD, which includes paired box gene eight (PAX8), TTF-2, NKX2.1 and NKX 2.5. Seven members of a non-consanguineous hypothyroid family with autosomal dominant mode of inheritance studied by Grasberger et al. had striking variable presentation. The proband and her brother who had elevated TSH and low free T4 on neonatal screening had normally placed gland on scintigraphy. At age of 37 years, their mother was found to have mild hypothyroidism on routine investigation. A female cousin had athyreosis along with elevated TSH and very low freeT4 on neonatal screening. Her five-year brother found to have elevated TSH and normal free T4 while their father was a hypothyroid since five years of age. A 67-year-old grandmother of the cousins had moderate thyroid failure on biochemical study. Sequence analysis revealed heterozygosity for PAX8 gene mutations among affected family members [9]. In this case report, we describe a family with five siblings who are diagnosed to have CH due to TD. This is supposed to be the family with maximum number of affected individuals reported so far in the medical literature. All siblings had athyreosis as a cause of CH. Mother is also hypothyroid with normally situated thyroid gland. Although common unidentified environmental factors cannot be ruled out, the involvement of genetic factor strongly suggested. Although genetic analysis could not be done in the present case but there is affair chance of PAX gene mutation responsible for CH as PAX mutation is autosomal dominant with incomplete penetrance. | ||||||
|
CONCLUSION
| ||||||
|
Familial congenital hypothyroidism (CH) due to thyroid dysgenesis is a rare though an important cause of permanent CH. Proper genetic counseling and universal screening for CH can prevent devastating later consequences. | ||||||
|
REFERENCES
| ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions
Abhinav Kumar Gupta – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Syed Mohd. Razi – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Final approval of the version to be published Deepak Chand Gupta – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Final approval of the version to be published Saqib Ahmad Khan – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Final approval of the version to be published Pankaj Jain – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Final approval of the version to be published Keshav Kumar Gupta – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Final approval of the version to be published |
|
Guarantor
The corresponding author is the guarantor of submission. |
|
Source of support
None |
|
Conflict of interest
Authors declare no conflict of interest. |
|
Copyright
© 2017 Abhinav Kumar Gupta et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information. |
|
|