
|
 |
|
Case Report
| ||||||
| A case of sudden cardiac arrest in a young adult | ||||||
| Zaid B. Al Jebaje1, John Elibol1, Robbie Wall1, Osman Saleem1 | ||||||
|
1Department of Internal Medicine, University at Buffalo, Catholic Health Systems, NY, USA
| ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article] [Similar article in Pumed] [Similar article in Google Scholar] 
|


| How to cite this article |
| Al Jebaje ZB, Elibol J, Wall R, Saleem O. A case of sudden cardiac arrest in a young adult. Int J Case Rep Images 2017;8(12):811–816. |
|
ABSTRACT
| ||||||
|
Introduction: Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited myocardial condition that primarily affects the right ventricle. The hallmark characteristic of this disease is continual loss and replacement of normal myocardium with fibrofatty tissue. This replacement of tissue can lead to life-threatening arrhythmias and potentially sudden cardiac death (SCD). Current diagnostic modalities include, electrocardiography, family history, echocardiogram, MRI scan, angiography and myocardial biopsy. Keywords: Arrhythmia, Arrhythmogenic right ventricular cardiomyopathy, Cardiomyopathy, Sudden cardiac arrest, Sudden cardiac death | ||||||
|
INTRODUCTION
| ||||||
|
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited myocardial condition that primarily affects the right ventricle. The prevalence of ARVC is approximately 1 in 2000 to 1 in 5000 in the general population and generally affects men more than women at a ratio of 3:1 [1][2]. The hallmark characteristic of this disease is continual loss and replacement of normal myocardium with fibrofatty tissue [3]. This replacement of tissue can lead to life-threatening arrhythmias and potentially sudden cardiac death (SCD). Despite the name, variations of ARVC can also affect the left side of the heart. In some cases, left-sided arrhythmias as well as left-sided heart failure may occur. Approximately 30–50% of cases of ARVC are considered familial and can be inherited in autosomal dominant or autosomal recessive forms. However, the autosomal dominant form is more common [4]. The pathogenesis is believed to be linked to over 30 genes that code for proteins involved in desmosomal structures between cardiac cells. The most known affected proteins are desmoplakin (DSP) [5], plakophilin 2 (PKP2) [6], desmoglein 2 (DSG2) [7], and desmocollin 2 (DSC2) [8]. These mutations cause a weakening in cell to cell mediated adhesions within the myocardium and lead to fibro-fatty replacement of normal ventricular tissue. Consequently, intense physical activity is restricted in these patients because of the increased risk of arrhythmia and sudden cardiac arrest/death. What makes this diagnosis challenging is that many of the patients suffering from ARVC are young, athletic individuals, who seem overtly healthy. The most common presenting symptoms can include syncope, cardiac ischemia, arrhythmia, sudden cardiac arrest or sudden cardiac death (SCD). The original diagnostic criteria, which was established in 1994, was revised in 2010 (Table 1). In 1994, a diagnosis of ARVC had to include either 2 major, 1 major + 2 minor, or 4 minor features. When the criteria were revised in 2010, three categories were introduced. They included, definite (2 major or 1 major + 2 minor), borderline (1 major + 1 minor or 3 minor), or possible (1 major or 2 minor) features for the diagnosis of ARVC. The purpose of this revision was to increase the sensitivity of the criteria and to increase the likelihood affected family members being identified before a cardiac event. The criterion was also revised in part to change in genetic testing and technological advances in medicine. If a patient survives the initial event, current diagnostic modalities include electrocardiography, family history, echocardiogram, magnetic resonance imaging scan, angiography and myocardial biopsy. | ||||||
| ||||||
|
CASE REPORT
| ||||||
|
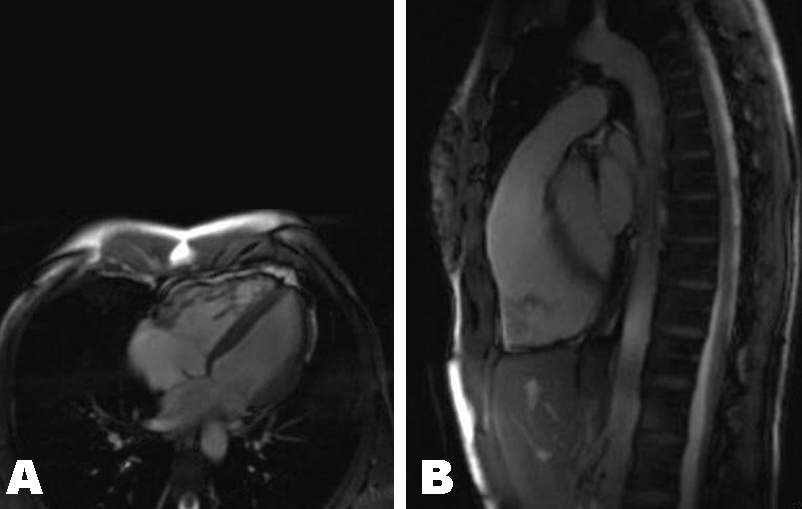
A 27-year-old athletic female with no known past medical history collapsed while playing frisbee in the park. Upon emergency medicine services (EMS) arrival, the patient was unconscious, pulseless, and in ventricular fibrillation. Cardiopulmonary resuscitation efforts were initiated and lasted 30 minutes. The patient received 8 DC shocks as well as on-scene intubation. Resuscitation attempts were successful and the patient was brought to the emergency department and transferred to the ICU. Initial electrocardigraphy (Figure 1) showed a QT interval of 460 ms and T-wave inversion in V1, V2, and V3. Transthoracic echocardiogram revealed a left ventricular ejection fraction (LVEF) of 30% along with moderate enlargement and reduced function of the right ventricle. The patient was successfully extubated four days after admission. Physical examination at that time was unremarkable. Follow-up transthoracic echocardiogram revealed a preserved LVEF of 55%. Cardiac catheterization was negative for coronary disease. The initial read of the cardiac MRI scan focused specifically on the left ventricle and was unremarkable. Genetic testing showed the patient was heterozygous for a novel variant of uncertain significance in the DSC2 gene that codes for desmosomal protein desmocollin-2, the pathogenic variants of which are found in autosomal dominant forms of ARVC. After the genetic study was performed, cardiac MRI scan with and without contrast (Figure 2) was read for a second time with a focus on the right ventricle. This revealed mild dilatation of the right ventricle, an elevated right ventricular end diastolic volume index of 111 mL/m2 (normal range 60–100 mL/m2), a decreased right ventricular ejection fraction (RVEF) of 36% (normally 60%), and no evidence of regional wall defects. Electrocardiography changes as well as the mutation in the DSC2 gene are consistent with two of the major criteria of ARVC and support a definitive diagnosis. Management at this time included a wearable defibrillator for 30 days, b-blockers, and abstaining from moderate and severe physical activity. The patient then received a single chamber subcutaneous intracardiac device (ICD) and was counseled on avoiding strenuous physical exertion. Six months later, she received an implantable ICD and has been following-up with our cardiac clinic. The patients first degree family members were all offered screenings. | ||||||
| ||||||
|
| ||||||
|
DISCUSSION
| ||||||
|
The diagnosis of arrhythmogenic right ventricular cardiomyopathy can be extremely challenging. This is because many patients are young and healthy adults before a symptomatic event ensues, usually precipitated by exercise. Unfortunately, by the time an event occurs, it may be too late because the most common presenting symptoms are cardiac ischemia, syncope, arrhythmia, or sudden cardiac arrest/death. ARVC accounts for 17% of cases of sudden cardiac death in the United States [9]. By definition our case represented a definitive diagnosis of ARVC due to two characteristics of the major criteria being met. This included T-wave inversion in V1–V3 with absence of right bundle branch block and mutation in desmocollin-2, which has a genetic association with ARVC [8][10]. Restricting strenuous physical activity is imperative for both affected individuals as well as healthy genetic carriers in order to prevent exercise induced cardiac events and progression of disease [11][12]. Additionally, clinical surveillance every two years and abstinence from intense physical activity is recommended in individuals with either unknown genotypes or in individuals who are known carriers of the diseases. Although there is no cure for ARVC, the name of the game is symptom management and prevention of arrhythmia or sudden cardiac death. This can be achieved with medical therapy that includes beta-blockers, diuretics, ace-inhibitors or angiotensin II receptor antagonists. Other therapies include endo/epi cardiac ablation or an intracardiac device. If these regimens prove to be suboptimal in symptom control, cardiac transplant is an alternative option. Ultimately, ARCV is a serious and complex cardiomyopathy that can result in ventricular arrhythmias and sudden cardiac death. The ARVC has an underestimated prevalence and is a challenging diagnosis that requires a high index of suspicion. Although many patients including the one discussed in this case do not show evidence of right ventricular damage, eventually all patients will. As such, diligent follow-up is imperative when treating patients with ARVC. Many aspects of ARVC have yet to be discovered and unfortunately no cure has been found to date. | ||||||
|
CONCLUSION
| ||||||
|
This case demonstrates the complex workup involved in cases of (ARVC) as well as the therapeutic options for these patients. This case also highlights the importance of counseling, affected patients and unaffected carriers, as well as screening of first-degree relatives in hopes of preventing serious unwanted outcomes. | ||||||
|
REFERENCES
| ||||||
| ||||||



|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions
Zaid B. Al Jebaje – Substantial contributions to conception and design, Acquisition of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published John Elibol – Substantial contributions to conception and design, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Robbie Wall – Substantial contributions to conception and design, Acquisition of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Osman Saleem – Substantial contributions to conception and design, Acquisition of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published |
|
Guarantor of Submission
The corresponding author is the guarantor of submission. |
|
Source of Support
None |
|
Conflict of Interest
Authors declare no conflict of interest. |
|
Copyright
© 2017 Zaid B. Al Jebaje et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information. |
|
|