|
|
|
|
Case Report
| ||||||
| Meckel-Gruber syndrome in Togo: Prenatal ultrasound and computer tomography diagnosis | ||||||
| Amadou A.1, Agbangba K.A.1, Amoussou K.1, Douaguibe B.2, Watara G.1, Sonhaye L.1, Adjenou V.1 | ||||||
|
1Radiology Department of CHU Campus 2Gynecology and Obstetrics Department of CHU Campus | ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article] [Similar article in Pumed] [Similar article in Google Scholar] 
|
| How to cite this article |
| Amadou A., Agbangba K.A., Amoussou K., Douaguibe B., Watara G., Sonhaye L., Adjenou V. Meckel-Gruber syndrome in Togo: Prenatal ultrasound and computer tomography diagnosis. Int J Case Rep Images 2017;8(10):648–652. |
|
ABSTRACT
| ||||||
|
Meckel–Gruber syndrome (MGS) is a rare autosomal recessive lethal disorder involving multiple systems. It is characterized by occipital encephalocele, polycystic kidneys and post-axial polydactyly. We report a case of MGS with occipital meningocele, bilateral enlarged echogenic kidneys, polydactyly, and severe oligohydramnios. The diagnosis was made by antenatal ultrasound and computer tomography scan. Meckel–Gruber syndrome is a syndrome rarely described in Africa. Its diagnosis is based on classic triad anomaly. None of this triad seems to be constant. Keywords: Meckel–Gruber syndrome, Ultrasound, Computer tomography, Togo | ||||||
|
INTRODUCTION
| ||||||
|
Meckel-Gruber syndrome (MGS) is a rare and lethal congenital polymorformative syndrome. It is a genetic disease, with autosomal recessive transmission, described for the first time in 1822 by Johan Friedrich Meckel [1]. It is characterized by the triad: encephalocele, cystic dysplasia of the kidneys and polydactyly [2]. Meckel-Gruber syndrome may also cause hepatic developmental defects, and pulmonary hypoplasia. Its global incidence ranges from 1 in 13,250 to 140,000 live births [3]. We report a case of MGS in Togo, whose antenatal diagnosis was made by ultrasound and completed by scanning. | ||||||
|
CASE REPORT
| ||||||
|
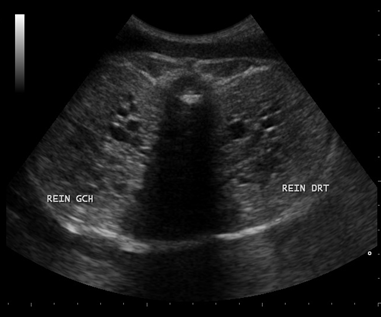
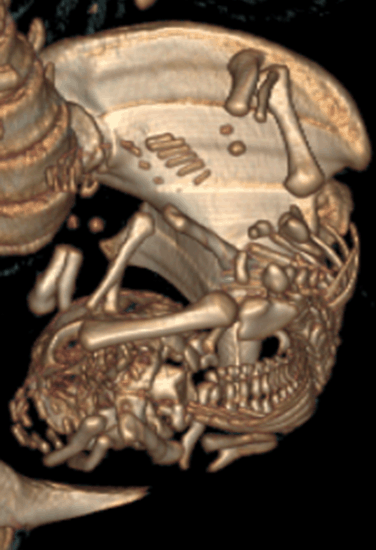
A 20-year-old female G1P0 presented to the obstetric department for the first time, with history of 28 weeks amenorrhea for routine antenatal examination. There was history of third-degree consanguineous marriage. She had her antenatal examination at a peripheral hospital. She was taking iron and folic acid (dosages of iron and folic acid were normal). Ultrasound done at our hospital as a routine, revealed a fetus with occipital meningocele (Figure 1), bilateral enlarged echogenic kidneys (Figure 2), polydactyly, and severe oligohydramnios. We found no liver abnormalities. For the precise analysis of polydactyly, a CT scan of the uterine contents was made. It showed a postaxial polydactyly affecting the four extremities, with six fingers at each hand and six toes at each foot (Figure 3). The scanner also found an occipital bone defect from which the encephalon herniated. The family was counseled and after getting consent, termination of pregnancy was planned immediately at 28 weeks’ gestation. Post-abortion macroscopic examination revealed multiple congenital anomalies including occipital encephalocele, and post-axial polydactyly. | ||||||
| ||||||
| ||||||
| ||||||
|
DISCUSSION
| ||||||
|
The Meckel–Gruber syndrome, also called dysencephalia splanchnocystica [4], is a rare polymalformative syndrome. The incidence in Belgium and Finland ranges from 1/3,000–1/9,000, respectively [5]. In India, highest incidence is in Gujrati Indians (1 affected birth per 1,300) [6]. Its incidence is not known in Africa and more particularly in Togo. This is a first discovery in our country. Some cases would probably have gone unnoticed. A high risk (25%) of recurrence in subsequent pregnancies is an important point for consideration and stresses the need for prenatal diagnosis in expectant mothers in those families. Meckel–Gruber syndrome affects all races with males and females being equally affected [4][7]. Early, (11–14th week) prenatal ultrasonography is the best method to diagnose MGS [7]. Subsequent autopsy and molecular studies are confirmatory. In our case, ultrasonography found this affection at 28th week. Meckel–Gruber syndrome is associated with multiple anomalies. It is characterized by classic triad of polycystic dysplastic kidneys, occipital encephalocele (or other anomalies of the central nervous system) and polydactyly [2]. All these three anomalies (triad) were found in our case. According to Sergi et al. [8], polycystic kidneys were found in all cases (100%), occipital encephalocele in 90% of case and post-axial polydactyly in 83.3% of cases. Salonen et al. [5] found a constant association between cystic dysplasia and liver fibrosis, and concluded that these two abnormalities, together with any other abnormalities of the nervous system, suffice to diagnose MGS. Fraser et al. [9] argue that only cystic renal dysplasia is essential for diagnosis. Wrigth et al. [10] never observed cystic dysplasia. The lack of consensus for the diagnosis of MGS led some authors to conclude that none of the anomalies of the triad or liver fibrosis is essential for diagnosis. The CT scan was almost never used for the diagnosis of MGS in literature. However, it can allow bone analysis in cases where the ultrasound is insufficient and the MRI scan is not available. In our case, the CT scan allowed to diagnose polydactyly and occipital defect. Meckel–Gruber syndrome has to be differentiated from other syndromes. The most likely syndrome to be confused with MGS is trisomy 13 [3]. Although the dismal outcome is the same for both, the recurrence rate is different. Trisomy 13 is mostly sporadic with low recurrence rate whereas MGS has 25% recurrence rate. Other syndromes similar to MGS are trisomy 18, Joubert syndrome, Bardet–Biedl syndrome and Smith–Lemli–Opitz syndrome [3][11]. The post-mortem assessment confirms the diagnosis, while genetic studies contribute to the evaluation of the recurrence risk [2][11]. Given the lethality of MGS in the perinatal or early infantile period, termination of pregnancy should be discussed if available [3]. | ||||||
|
CONCLUSION
| ||||||
|
Meckel-Gruber syndrome is a syndrome rarely described in Africa. Its diagnosis is based on classic triad anomaly. None of this triad seems to be constant. The isolation of the gene responsible for this syndrome would therefore be necessary for a diagnosis of certainty. | ||||||
|
REFERENCES
| ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions
Amadou A. – Substantial contribution to conception and design, Analysis and interpretation, Drafting the article, Final approval of version to be published Agbangba K.A. – Substantial contribution to conception and design, Analysis and interpretation, Drafting the article, Final approval of version to be published Amoussou K. – Substantial contribution to conception and design, Analysis and interpretation, Drafting the article, Final approval of version to be published Douaguibe B. – Substantial contribution to conception and design, Analysis and interpretation, Drafting the article, Final approval of version to be published Watara G. – Substantial contribution to conception and design, Analysis and interpretation, Drafting the article, Final approval of version to be published Sonhaye L. – Substantial contribution to conception and design, Analysis and interpretation, Drafting the article, Final approval of version to be published Adjenou V. – Substantial contribution to conception and design, Analysis and interpretation, Drafting the article, Final approval of version to be published |
|
Guarantor
The corresponding author is the guarantor of submission. |
|
Source of support
None |
|
Conflict of interest
Authors declare no conflict of interest. |
|
Copyright
© 2017 Amadou A. et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information. |
|
ABOUT THE AUTHORS
| |||||||||||||||||||||
| |||||||||||||||||||||
|
|