  |

|
 |
|
Clinical Image
| ||||||
| An unusual case of Erdheim–Chester disease | ||||||
| Guillermo Andres Cortes1, Sara C. Acree2, Helga Van Herle1 | ||||||
|
1Department of Medicine, Division of Cardiology, University of Southern California/Los Angeles County Medical Center.
2Department of Pathology, University of Southern California/Los Angeles County Medical Center. | ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article]
[Similar article in Pumed] [Similar article in Google Scholar] 
|


| How to cite this article |
| Cortes GA, Acree SC, Herle HV. An unusual case of Erdheim–Chester disease. Int J Case Rep Images 2016;7(9):606–611. |
|
Case Report
| ||||||
|
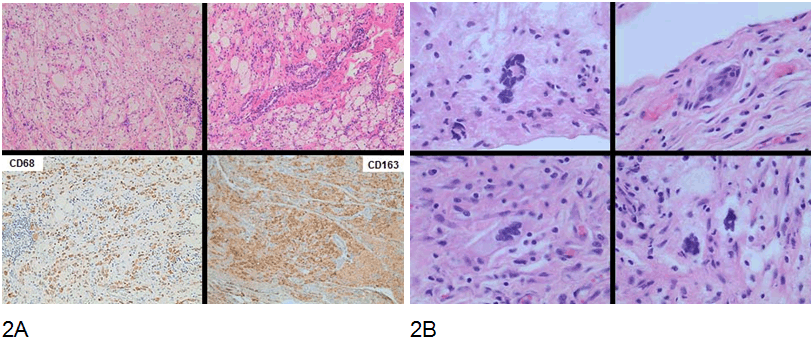
A 29-year-old female with a two-year history of central diabetes insipidus and hypothyroidism presented with progressive diffuse abdominal pain. A computed tomography (CT) scan of the abdomen (Figure 1) revealed diffuse infiltrative disease likely fibrosis. Her abdominal pain worsened over time and she underwent an exploratory laparotomy, which revealed an extensive process resembling severe fibromatosis versus carcinomatosis. Due to marked inflammation surrounding the appendix and right ovary, the patient underwent appendectomy and right oophorectomy. In addition, multiple biopsies were taken of the peritoneum and omentum (Figure 2A-B). The pathologic diagnosis was reported as a fibrohistiocytic process. The patient's postoperative course was complicated by bilateral hydronephrosis. Cystoscopy and ureteroscopy with bilateral ureteral stent placement was performed with no improvement. Ultimately required bilateral nephrostomy tube placement; renal function continued to deteriorate and she was started on hemodialysis. Shortly thereafter, she developed a deep venous thrombosis of the left upper extremity and anticoagulation with warfarin was instituted. At the same time, the patient developed severe abdominal pain. A CT scan of the abdomen revealed a large bowel obstruction requiring surgical intervention with partial colectomy and colostomy. The patient was subsequently transferred to our institution for further evaluation and higher level care. Two weeks after anticoagulation was started, the patient developed bleeding from the colostomy site. However, she remained hemodynamically stable. The gastroenterology service evaluated the patient and determined that the risk of perforation from an EGD and colonoscopy to find the source of bleeding or from performing a new biopsy was significantly high. It was further determined that the site of bleeding was at the colostomy stoma. As the patient's primary diagnosis remained unclear and CT images were reviewed. A mass at the inferior vena cava right atrium was identified (Figure 3). The initial transthoracic echocardiogram demonstrated a dimension 2.5x2.4 cm, non-mobile mass, extending almost from the entrance of the IVC to the posterolateral wall of the right atrium (Figure 4). An MRI scan was attempted however, patient, did not tolerate the study and it was aborted, the few images captured did demonstrate evidence of the mass (Figure 6). A cardiac CT scan allowed us to have a better visualization of the mass. It was found to be 3.9x3.8 cm in dimension, intraluminal within the right atrium, centered on the crista terminalis and infiltrating into the right atrial-ventricular groove and the intra-atrial septum (Figure 5). A cardiac biopsy was performed and revealed a process not unlike what was seen in the abdomen. This tumor was comprised of bland-appearing fibroblast like cells characterized as medium-sized, with oval to stellate nuclei, fine chromatin, inconspicuous nucleoli, and ample amphophilic to eosinophilic cytoplasm with indistinct cell borders. These cells were scattered within a myxoid to histiocytic background. Like the intra-abdominal tumor, these cells were immunoreactive for CD68 and CD163 (Figure 2) and (Figure 7). These histologic findings, combined with the clinical presentation, was most consistent with non-Langerhans cell histiocytosis, otherwise known as Erdheim–Chester disease. Bone scan was performed and no evidence of osteosclerosis or infiltrative disease was found (Figure 8). The patient's clinical course had been most complicated by the aggressive peritoneal and retroperitoneal proliferation, which had caused bowel obstruction and ureteral obstruction, with hydronephrosis and renal failure, and cardiovascular infiltrating mass extending from IVC into the right atrium with no hemodynamic compromise. However, after initiation of therapy with Anakinra, a recombinant IL-1 receptor antagonist; she showed a dramatic clinical improvement. Ultimately, the patient responded well to therapy and was send home with plan to follow as outpatient. | ||||||
| ||||||
|
| ||||||
| ||||||
| ||||||
|
| ||||||
| ||||||
|
| ||||||
|
| ||||||
|
| ||||||
|
Discussion
| ||||||
|
Jakob Erdheim and William Chester first described the "Über Lipoidgranulomatose" in 1930 [1]. The term "Erdheim–Chester disease" (ECD), however, was first used by Elaine Jaffe (1972) [2] [3]. Erdheim–Chester disease is a rare, non-Langerhans form of histiocytosis [2] [4][5] [6][7]. It is an adult variant of the disseminated juvenile xanthogranulomatosis, characterized by a proliferation and multiorgan infiltration of histiocytes [1]. Histologically, this disorder is characterized by a relatively bland-appearing epithelioid to spindle cell proliferation often with a foamy (xanthomatous) component, especially in older lesions, and variably-prominent multinucleated Touton-type giant cells (xanthogranulomatous). This histiocytic proliferation is positive for histiocytic markers, such as CD68 and CD163, and negative for CD1a and Langerin [2] [4][6][8]. In most cases (80%), staining for S-100 protein is also negative [2][4] [5][6][9][10]. Erdheim–Chester disease (ECD) must be distinguished from Langerhans cell histiocytosis (LCH), which includes three subtypes, the unifocal eosinophilic granuloma, the multifocal unisystem Hand-Schuller–Christian disease and the multifocal, multisystem Letterer-Siwe disease. Classically, LCH presents with asymmetric lytic lesions of flat bones, whereas ECD is associated with bilateral, symmetric diffuse metaphyseal and diaphyseal cortical osteosclerosis of long tubular bones of the appendicular skeleton with sparing of the epiphyses [7] [11]. Immunohistological, LCH stains positive for S-100 protein, CD1a and Langerin. Electron microscopy reveals the characteristic "tennis-racket" cytoplasmic organelles called Birbeck granules, also known as X bodies, whose function is not entirely known [5] [7]. In contrast, ECD is negative for CD1a and Langerin and does not have these ultra-structural findings. Therefore, ECD is classified as a non-Langerhans cell histiocytosis. Evans et al. have called it "polyostotic sclerosing hystiocytosis" [11]. Clinical features of ECD range from asymptomatic presentation to extensive multiorgan infiltration and death [2][4][5][10]. Bone pain is the most common manifestation, seen in up to 50% of the cases. More than half of the patients with ECD have extra skeletal involvement, including neurological (exophthalmos, gaze disturbances, ataxia), diabetes insipidus, hypogonadism, hypopituitarism from hypothalamic infiltration, retroperitoneal involvement (renal failure, hydronephrosis, nephrovascular hypertension), dyspnea (lung and pleural involvement), cutaneous manifestations (xanthoma, xanthelasma) and cardiovascular involvement, (seen in up to 60% of cases [1] [2][4][5][6][8] [10][11][12]. Haroche et al. were the first to look at the cardiovascular involvement in ECD. In 2004, they published a literature review that concluded significant underestimation in the extent of cardiac involvement. Of the 178 cases of ECD reviewed, 72 cases had CV involvement: 56% had periaortic fibrosis and 28% presented with fibrosis of the aorta (coated aorta) [2] [13]. Direct heart involvement was found in 54 cases; this involvement included pericardial infiltration (44%), myocardial infiltration (31%), valvular heart disease (8%), a right atrial tumor (8%). Heart failure was seen in 19 cases with death resulting in eight cases; myocardial infarction occurred in six cases including two deaths. Cardiovascular complications were responsible for the death in 31.4% of the cases [2] [10], confirming the poor prognosis of patients with CV involvement in ECD [2][4][5] [6][10][13]. In 2009, Haroche et al. report the evaluation of 37 patients with cardiac magnetic resonance imaging scan and computer tomography. Of these, 26 patients (70%), had abnormalities including direct infiltration of right heart (49%), infiltration of the left coronary artery (27%), pericardial effusion (24%), and pericardial thickening (14%) [8]. In recent years, B-Raf V600 mutations have been described in various malignancies, including melanoma, colorectal and thyroid carcinomas, hairy cell leukemia, and histiocytosis. In the latter group, which includes both Erdheim-Chester disease and Langerhans cell histiocytosis, this mutation has been identified in roughly half the cases [9]. B-Raf is a member of the Raf kinase family of growth signal transduction protein kinases and plays a role in regulating the MAP kinase/ERKs signaling pathway, which affects cell division, differentiation, and secretion. Testing for this mutation has become critical with the availability of vemurafenib, which stands for "V600E mutated BRAF inhibition". As an inhibitor of mutated BRAF, vemurafenib interrupts the B-Raf/MEK/ERK pathway, and it was initially FDA-approved for the treatment of late-stage melanoma. More recently, however, it has been shown to be effective in the treatment of histiocytoses [14]. This patient was tested for the BRAF mutation, but because she was negative, she was not treated with vemurafenib, but instead with anakinra. Anakinra is a recombinant IL-1 receptor antagonist and appears over-stimulated in ECD. IL-1 receptor antagonist synthesis is naturally induced after stimulation of IFN-alpha, which has been shown to be increased in ECD [15]. | ||||||
|
Conclusion
| ||||||
|
In summary, Erdheim–Chester disease (ECD) is multisystem infiltrative disease of histiocytic origin. It is imperative to screen patient with ECD for possible cardiovascular involvement as was seen in this patient, as cardiac involvement denotes a much more aggressive form of this disorder. Cardiac MRI and CT scans are valuable complements to echocardiography for visualization and characterization of cardiac and pericardial structures in order to direct the most appropriate therapy. Still is unclear if the treatment affects the overall course of the patients with cardiovascular involvement of ECD. Keywords: Erdheim-Chester Disease, Aggressive fibromatosis, Disseminated juvenile xanthogranuloma | ||||||
|
References
| ||||||

| ||||||







|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions
Guillermo Andres Cortes – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Sara C. Acree – Analysis and interpretation of data, Revising it critically for important intellectual content, Final approval of the version to be published Helga Van Herle – Analysis and interpretation of data, Revising it critically for important intellectual content, Final approval of the version to be published |
|
Guarantor of submission
The corresponding author is the guarantor of submission. |
|
Source of support
None |
|
Conflict of interest
Authors declare no conflict of interest. |
|
Copyright
© 2016 Waqas Jehangir et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information. |
|
|