  |

|
 |
|
Case Report
| ||||||
| Determining primary from secondary hyperparathyroidism: A review of current management | ||||||
| Michael Smith1, Sherwin Schrag2 | ||||||
|
1DO, Department of Surgery, St Barnabas Hospital, Bronx, NY.
2MD, Department of Surgery, Jersey City Medical Center, Jersey City, NJ. | ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article]
[Similar article in Pumed] [Similar article in Google Scholar] 
|


| How to cite this article |
| Smith M, Schrag S. Determining primary from secondary hyperparathyroidism: A review of current management. Int J Case Rep Imag 2016;7(6):365–369. |
|
Abstract
|
|
Introduction:
Primary hyperparathyroidism is the third most common endocrine disorder and the most common cause of hypercalcemia. Since the advent of automated serum chemistry machines, it is more common for hypercalcemia to be discovered incidentally. Secondary hyperparathyroidism is seen in the setting of chronic renal insufficiency. Both result in elevated parathyroid levels and in a patient with underlying renal disease distinguishing the primary from the secondary disease may be difficult.
Case Report: A 60-year-old female presented to an emergency department with the complaints of constipation, weakness, dizziness that began acutely. She began to receive routine renal replacement therapy one month prior to presentation. A serum calcium level of 15 mg/dl was found on routine blood work. Further workup revealed a parathyroid hormone level of over 1000 pg/mL. Ultrasonography of the neck and sestamibi scan confirmed the location of the suspected lesion. On removal of the mass, which histologic examination revealed to be a 6.2 grams parathyroid adenoma, her calcium level returned to normal. Conclusion: Primary hyperparathyroidism requires surgical intervention for definitive therapy. Secondary hyperparathyroidism requires optimization of medical therapies to treat effectively in lieu of renal transplantation. In patients with underlying renal insufficiency and elevated calcium levels, imaging should be used to rule out parathyroid adenomatous disease and parathyroid carcinoma. | |
|
Keywords:
Parathyroid adenoma, Parathyroid carcinoma, Primary hyperparathyroidism, Secondary hyperparathyroidism
| |
|
Introduction
| ||||||
|
Primary hyperparathyroidism affects 0.1–1% of patients and 0.3 per 1000 annually of the population, most commonly found incidentally on chemistry panels. The traditional pentad of bony, renal, gastrointestinal, neuropsychiatric and neuromuscular fatigue has fallen by the wayside due to advances in medical technology [1][2]. An unusual presentation of hyperparathyroidism is the giant parathyroid adenoma, a hyperfunctional and hypertrophic response of the parathyroid glands. Primary and secondary hyperparathyroidism have separate pathological processes which both lead to bony destruction and elevated serum calcium with its resultant sequelae [3] [4]. Primary and secondary hyperparathyroidism may occur in patients with parathyroid adenoma and chronic renal disease, as we intend to discuss fully, and can muddle the diagnosis of the former [5] [6][7]. | ||||||
|
Case Report
| ||||||
|
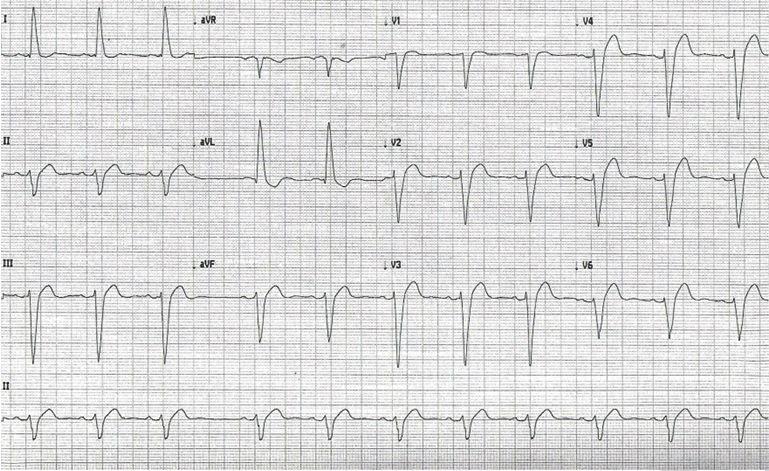
A 60-year-old female presented to Jersey City Medical Center after a several hour course of palpitations. Believing this to be caused by hypertension, she took her prescribed metoprolol, hydralazine, and clonidine at that time in an attempt to treat. No symptoms of chest pain, shortness of breath, or lightheadedness were described. She returned to sleep but awoke several hours later feeling lightheaded and weak. She attempted to walk from her bedroom, but due to this malaise and dizziness she laid down on the floor. She had secondary complaints of constipation denying a bowel movement for six days, although continued to pass flatus. Medical history of the patient is significant for end stage renal disease, having recently started hemodialysis one month prior to presentation, hypertension, diabetes, left bundle branch block, morbid obesity, hypothyroidism, gout, gastroesophageal reflux disease and bilateral renal cell carcinoma. Surgical history was significant for a left nephrectomy, a right partial nephrectomy, and tunneled hemodialysis catheter insertion into the right internal jugular vein. On arrival to the emergency department, she was bradycardic at 52 beats per minute. An electrocardiogram was performed, showing sinus bradycardia at 45 beats per minute, widening of the QRS complex with markedly deepened S waves, a significantly shortened QT interval, and LBBB (Figure 1). Standard lab investigations showed showing elevated calcium levels of 15.6 mg/dL and ionized calcium of 1.98 mg/dL. Bisphosphonate (alendronate 10 mg by oral administration daily) and calcimimetic (calcitonin 200 mg intranasal administration daily) therapy was begun on admission. Approximately one month prior to admission, she had an intact parathyroid hormone (PTH) level of 1046 pg/mL. A sestamibi scan was performed and the left inferior parathyroid was found to be hyperactive. An ultrasound was also performed in the outpatient setting and the patient was found to have a complex mid-pole nodule measuring 2.8x2.1x2.1 cm. Due to the symptomatology of the parathyroid adenoma, the patient was prepared for surgery. The morning prior to surgery, the patient's PTH level was 1496 pg/mL. A minimally invasive parathyroidectomy was performed. Pathologic examination revealed a 6200 mg parathyroid adenoma. Following the procedure, calcium levels returned to normal. | ||||||
| ||||||
|
Discussion
| ||||||
|
In a non-pathologic state, 99% of the normal calcium resides in bone and the remaining 1% is rapidly exchanged with the nonosseous extracellular calcium pool. Of the calcium found intravascularly, 50% of total calcium is bound to the serum proteins, albumin and the globulins. The intracellular calcium is mainly found in the mitochondrial compartment, bound to the inner plasma membrane or with proteins in the endoplasmic reticulum. Parathyroid hormone, PTH, controls the minute to minute level of calcium within the serum. It is a protein composed of 84 amino acids derived from a larger, pre-pro-parathyroid hormone. After cleavage of the precursor amino acid sequences, the mature hormone is sequestered in secretory vesicles and granules. In the setting of hypercalcemia, under normal conditions, the release of PTH is substantially decreased and what is released is an inactive carboxy-terminal fragments. The PTH stimulates the release of calcium by acting on osteoclasts to increase in number and activity in addition to resorption by the kidneys [1]. With the advent of automated blood chemistries in the 1970s, the traditional pentad of "bones, psychic moans, stones, abdominal groans, fatigued overtones" has fallen by the wayside as incidental hypercalcemia has become more prevalent [2] [8]. Kidney stones occur in only 10–25% of patients although one-third of patients have a reduction in creatinine clearance or impaired concentrating or acidifying ability may occur [1]. Less than 5% of primary hyperparathyroidism (pHPT) patients have skeletal involvement since this advent [8]. The reported prevalence of hypercalcemia in patients ranges from 0.1–1% with an estimated annual incidence of 0.3 per 1000 persons with a ten-fold increase in those aged 15 to 65 years (0.7 to 15 per 1000) in the United States [1] [9]. Osteitis fibrosa cystica and brown tumors, or giant cell fibroblastic tumors filling osteolytic defects, are typically the presentation of skeletal involvement from pHPT [8]. Brown tumors usually occur in the medullary shaft of long bones, but also include the mandible, maxilla, clavicle, metacarpal bones, ribs, pelvic bones, and rarely the cranium. Multiple brown tumors are exceedingly rare, only six cases reported in English literature [10]. Histologically, these tumors are non-encapsulated and are characterized by abundant vascularity, stroma consisting of fibrous connective tissue, with a proliferation of fibroblasts, deposition of hemosiderin. Radiographically, these tumors show widespread loss of lamina dura and the subperiosteal erosion of the phalanges. Computed tomography reveals a lytic lesion either hyperdense or heterogeneous with associated soft tissue mass either well-circumscribed or expansile lucent lesion with a rim of calcification and remodeling of the surrounding bone. On magnetic resonance imaging, a hypointense signal is found on T1 weighted imaging, variable signal on T2 weighted images and loss of signal on in-phase images because of the magnetic susceptibility from hemosiderin [10]. Primary hyperparathyroidism is the most common cause of hypercalcemia and the third most common endocrine disorder, after diabetes and hypothyroidism [11]. The pathogenesis results most often from a solitary parathyroid adenoma 75–85% of the time. In 15–20% two or more adenomas are found and in one percent parathyroid carcinoma is discovered [2] [10][12]. Primary hyperparathyroidism results from a defect in proliferation leading to hypercellularity of one or more glands but also a defect in the negative feedback system [13]. Mean preoperative PTH levels of 180 pg/mL is common in pHPT while levels greater than 700 pg/mL indicate parathyroid carcinoma [3]. Only sixteen cases of parathyroid adenoma larger than 30 grams have been documented as of 2011. The largest parathyroid adenoma recorded was 8x5x3.5 cm and weighed 110 grams [14]. A single gland above 60 milligrams is unusual and the usual weight of a parathyroid adenoma ranges from 70 mg to 1 g [2]. Tumors larger than two to five grams are deemed giant parathyroid adenomas based on the local definitions [11] [14]. One to two percent of patients with pHPT present with parathyroid crisis, a life-threatening condition in which severely elevated calcium levels lead to multiorgan failure [3]. The bony manifestations of classic hyperparathyroidism include osteitis cystica fibrosa caused by over absorption of bone by osteoclastic stimulation with radiographic evidence of demineralization of bone and coarsening of the trabeculae. In terms of cardiovascular physiology, the increased calcium leads to a shortened QT interval. In terms of renal symptomatology, recurrent nephrolithiasis, nephrocalcinosis or impaired concentrating ability to end stage renal disease can occur from pHPT. Less common symptoms include anorexia, pancreatitis, peptic ulcer disease, band keropathy, hypertension. Elderly patients are especially prone to neuropsychiatric symptoms such as irritability, emotional instability, fatigue, apathy, depression, dementia and psychosis [1]. Secondary hyperparathyroidism (sHPT) is defined by increased secretion of parathyroid hormone in the setting of chronic renal disease due to the kidneys inability to secrete phosphate and perform the final conversion to metabolically active vitamin D. The increased secretion of PTH ultimately leads to the symptoms of primary hyperparathyroidism. Levels of PTH are severely elevated and may lead to the development of brown tumors of the jaw and long bones, although rarely seen in pHPT, is much more common in sHPT [7]. The work done on transgenic mice have shown that cyclin D1 oncogene overexpression is a key mechanism in both primary and secondary hyperparathyroidism. This mutation also induces a decrease in calcium receptors leading to increased production of hyperparathyroid hormone [13]. Overexpression of tumor necrosis factor alpha (TNFa) secondary to increased endothelial growth factor receptor (EGFR) expression has also been shown to be an important factor in the pathogenesis of secondary hyperparathyroidism. In mice undergoing nephectomy with resultant end stage renal disease, EGFR increase caused increased TNFa levels. This resulted in parathyroid hyperplasia and increased PTH production and release. After administering erlotinib, a monoclonal antibody potent and specific for EGFR led to decreased TNFa and thusly decreased PTH production and release [15]. In a 15-year prospective study, 59 patients with asymptomatic, incidentally found hyperparathyroidism were followed to evaluate the long term sequelae of their disease without intervention. Although the eight year bone mineral density (BMD) studies showed no change and the lumbar spine BMD showed no change over the course of the study. However, at the 10 year BMD study of the radius, one-third of the subjects showed accelerated losses. During the study 18 patients developed new surgical criteria during the study but this did not predict progressive disease [6]. Based on an extensive compilation of research, the Third international workshop on the management of asymptomatic hyperparathyroidism recommended cinacalcet as the primary treatment for this disease for the prevention of renal symptoms by reducing the release of PTH and effectively decreasing serum calcium levels. Selective estrogen receptor modulators (SERMS, i.e., raloxifene) did not show to be efficacious in reducing the serum calcium load. For those patients in which skeletal protection was the primary goal, bisphosphonates, such as alendronate, and hormone replacement therapy were shown to be equally effective [4] [5] . Cinacalcet, a calcimimetic, has been used in both primary and secondary hyperparathyroidism. Calcimimetics increase the sensitivity of calcium receptors leading to inhibition of parathyroid hormone levels within a few hours after administration [16]. Peacock reported on the results of 78 patients with pHPT half of whom were given cinacalcet in titrated doses for 12 weeks, a maintenance phase for another 12 weeks and followed for an additional 28 weeks. During the maintenance phase 73% of patients achieved the primary endpoint of serum calcium <10.3 mg/dL compared with 5% in the placebo group (p <0.001). The BMD was measured pretreatment, at 24 weeks and at 52nd week; at 24th week, the mean change in Z score at the lumbar spine was significantly lower in the cinacalcet group than the procedure (p <0.05) however no significant difference was observed at 52nd week [17]. Secondary hyperparathyroidism response to cinacalcet was evaluated in comparison to placebo in 741 patients with 12 weeks of titration from initial dose of 30 mg daily, 14 weeks of efficacy assessment. Increases were permitted if parathyroid hormone levels were greater than 200 pg/mL. Results from this study showed that cinacalcet treated patients had significantly lower parathyroid hormone levels (p <0.001) than the placebo group. Serum calcium and phosphorous were significantly decreased compared with the placebo group [16]. | ||||||
|
Conclusion
| ||||||
|
In conclusion, primary and secondary hyperparathyroidism develop from two separate pathological processes. Incidentally elevated hypercalcemia should be promptly evaluated for the cause. Symptomatic primary hyperparathyroidism should be managed surgically while asymptomatic cases may elect for surgical intervention or medical management. Secondary hyperparathyroidism demands medical management and aggressive correction of elevated serum calcium and phosphate levels. | ||||||
|
References
| ||||||
| ||||||

|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions
Michael Smith – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Sherwin Schrag – Analysis and interpretation of data, Revising it critically for important intellectual content, Final approval of the version to be published |
|
Guarantor of submission
The corresponding author is the guarantor of submission. |
|
Source of support
None |
|
Conflict of interest
Authors declare no conflict of interest. |
|
Copyright
© 2016 Michael Smith et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information. |
|
|