  |

|
 |
|
Case Report
| ||||||
| Compound heterozygous deletions presenting as infantile chylomicronemia | ||||||
| Susanna Felsenstein1, Geesje Dallinga-Thie2, Shankar Kanumakala3 | ||||||
|
1Great Ormond Street Hospital, London, United Kingdom.
2Academisch Medisch Centrum Universiteit van Amsterdam. Department of Vascular Medicine, Amsterdam, Netherlands. 3Royal Alexandra Children's Hospital, Brighton, United Kingdom. | ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article]
[Similar article in Pumed] [Similar article in Google Scholar] 
|


| How to cite this article |
| Felsenstein S, Dallinga-Thie G, Kanumakala S. Compound heterozygous deletions presenting as infantile chylomicronemia. Int J Case Rep Images 2015;6(11):712–716. |
|
Abstract
|
|
Introduction:
Monogenic disorders affecting the lipid metabolism are rare, but early diagnosis is important to ensure prompt initiation of management.
Case Report: A three-week old neonate presenting with rectal bleeding was found to have blood of an unusual cream-like appearance. Significant lipemia was confirmed, with massive increase in triglyceride and cholesterol levels. Centrifugation confirmed chylomicronemia. Primary chylomicronemia is extremely rare and most commonly caused by a lipoprotein lipase (LPL) gene mutation. The infant was a compound heterozygous for two deletions in the LPL gene: p. Thr45HisfsX3, and the here for the first time described p. Phe189X, both leading to a premature stop codon the absence of a mature protein. Subsequent change to medium chain triglyceride feed resulted in near-normal blood lipid levels. Conclusion: Novel mutations affecting chylomicron metabolism continue to be identified and may affect patients of ethnic background considered low-risk. This case illustrates that adequate treatment and dietary management is highly effective in symptomatic management, and in preventing serious complications in both infancy and later in life. | |
|
Keywords:
Child, Chylomicronemia, Hyperlipidemia, Infant, Lipoprotein lipase gene mutation, Neonate, Ultracentrifugation
| |
|
Introduction
| ||||||
|
Infantile presentation with gross hyperlipidemia is rare, and may pose a challenge to the front-line pediatrician in differential diagnosis, therapeutic management and follow-up [1]. The diagnosis will rarely be straightforward, as manifestations are not diagnostic, but can be as diverse and severe as liver failure, pancreatitis, coagulopathy or fat emboli formation including the possibility of infantile stroke or other vascular complications. Herein, we present a case of infantile chylomicronemia illustrating these points as well as reviewing current diagnostic and treatment concepts. | ||||||
|
Case Report
| ||||||
|
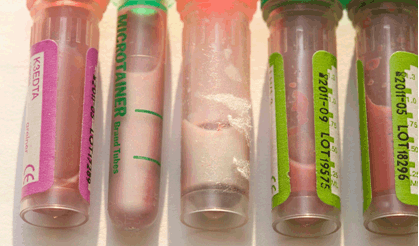
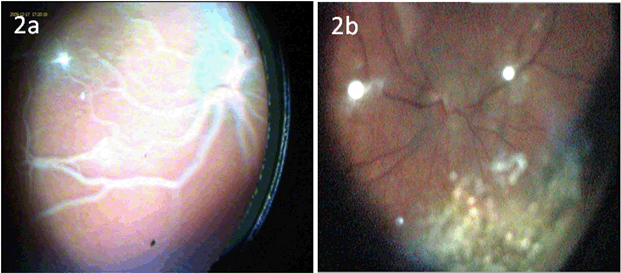
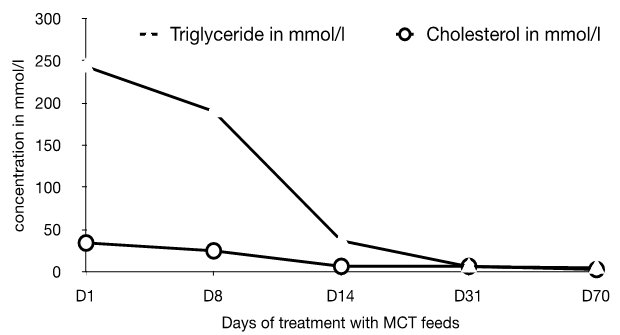
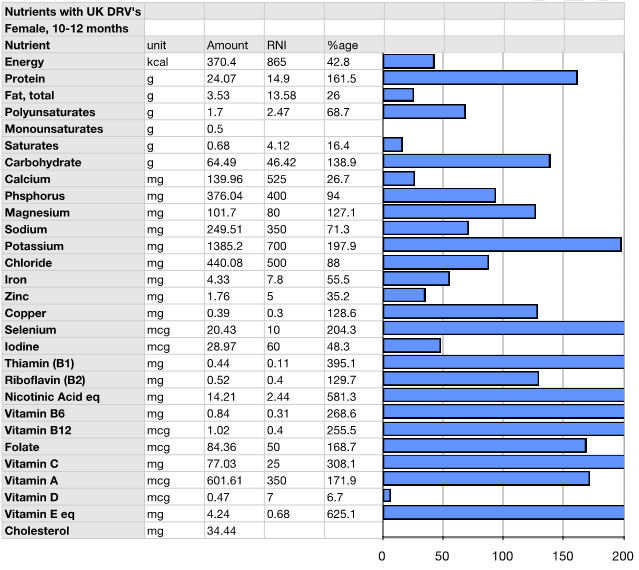
Presentation and Diagnostic workup Coagulation profile was normal, though results were difficult to obtain because of the severe hyperlipidemia. The initial fasting lipid profile showed a triglyceride level of 243.3 mmol/L (normal range: 0.8–1.7 mmol/L). Measurements taken of other blood lipids were not valid due to the degree of hypertriglyceridemia. Hemogram, renal and liver function tests including amylase were within normal range (hemoglobin 10.7 g/dL, white blood cell count 21.2x109/L, platelet 299x109/L, hematocrit 0.3, MCV 98 fL, MCH 35.1 pg, neutrophils 11.2x109/L, lymphocytes 7.7x109/L, monocytes 1.8x109/L, eosinophils 0.5x109/L. Na 142 mmol/L, K 5.0 mmol/L, urea 1.6 mmol/L, creatinine 27 umol/L, albumin 41 g/L, total protein 62 g/L, ALT 17 IU/L, blood glucose 5.3 mmol/L (normal range: 4.4–6.1 mmol/L), amylase 46 IU/L (normal range: 28–100 IU/L)). Thyroid function tests demonstrated a TSH of 2.39 mU/L (normal range: 0.3–4.2 mU/L), and free thyroxine of 17.5 pmol/L (normal range: 12–22 pmol/L), excluding hypothyroidism. A blood sample after three days of breastmilk feeds revealed near-normal lipid profile following removal of the chylomicron fraction by ultracentrifugation, establishing presence of chylomicronemia. A fundoscopy at this point illustrated classical lipemia retinalis (Figure 2A). Genetic work-up: The patient's father showed a slightly raised triglyceride level at 2.4 mmol/L (Reference: 0.8–1.7 mmol/L), her mother had marginally elevated cholesterol at 5.7 mmol/L (Reference range: 2.8–5.0 mmol/L). Both parents are thus clinically asymptomatic carriers of one recessive allele with mild derangement of their lipid profile. Management This diagnosis entails a lifelong condition, with complex management challenges throughout life. Through the first year of life the patient maintained near-normal levels of both Cholesterol and triglycerides (TGs) on Monogen® to date. The necessity of avoiding long chain fatty acids was expected to make weaning onto a solid diet and nutrition challenging and is therefore closely supervised by a dietician. Weaning has been successful so far and the patient is on to a largely fat free solid diet, rich in protein and carbohydrates. As a result of the strict restriction on fat intake, the infant receives 66% of her energy from protein, 26% from carbohydrate intake, and only 8% of her energy from fat, compared to the normally recommended 30% of energy derived from fat intake in this age group Table 1. Careful planning of the diet with additional Monogen® feeds ensures the patient's micronutrient requirements were met in infancy. Supplementation with Walnut oil (0.1 ml/56 kcal energy requirement) will provide the appropriate ratio of essential fatty acids. Close dietary monitoring will be necessary throughout to ensure all her macronutritional and micronutritional needs are met. Repeat fundoscopy at six months of age showed complete resolution of lipemia retinalis (Figure 2B). The patient's cholesterol and TG levels have successfully been maintained at near-normal levels following the introduction of a solid diet, and she continues to thrive and develop normally. | ||||||
| ||||||
| ||||||
| ||||||
| ||||||
|
Discussion
| ||||||
|
Primary chylomicronemia (also known as familial hypertriglyceridemia type I) is a rare autosomal recessively inherited disorder. The estimated prevalence rate of chylomicronemia is reported at around 1 in 1 million [1]. A founder effect results in a slightly higher prevalence in Quebecois Canadians. It is most commonly caused by a mutation in LPL, a key enzyme in TG metabolism located on the luminal surface of endothelium [2]. It aids the absorption of free fatty acids in peripheral tissues, catalyzing the hydrolysis of chylomicron-TGs and VLDL-TGs, thus delivering free fatty acids to peripheral tissues either for direct energy delivery or storage. Lack of or malfunction of LPL results in the inability to hydrolyze triglycerides from chylomicron and VLDL particles and leads to a massive rise in plasma levels of chylomicron and VLDL particles and accumulation of TGs in the plasma compartment [2] [3]. In this case, the patient was found to be heterozygous for two LPL mutations resulting in premature stop codons. LPL requires a cofactor, apolipoprotein CII (APOC2), which resides on TG-rich particles and on HDL. Deficiency in apolipoprotein CII (APOC2) is very rare, but affected patients develop a similar phenotype, as LPL cannot function properly resulting in absence of TG hydrolysis and accumulation of TGs in plasma. Two more proteins play an essential role in LPL homeostasis: Lipase Maturation Factor 1 (LMF1) and glycosylphosphatidylinositol (GPI)-anchored HDL binding protein 1 (GPIHBP1). Deficiencies in LMF1 and GPIHBP1 result in a similar clinical phenotype as LPL deficiency, with severe chylomicronemia at a young age [3] [4] [5]. LMF1 is an intracellular protein involved in the maturation of LPL. In the absence of functional LMF1 no active LPL will be secreted, again leading to severe hypertriglyceridemia similar to LPL deficiency. GPIHBP1 acts as the platform at the endothelial cell surface that provides LPL and TG rich lipoprotein particles to meet and subsequently allow TG hydrolysis [6]. The phenotype of these patients is similar to LPL deficiency. Deficiency of apolipoprotein A5 (APOA5) does not lead to hypertriglyceridemia but requires a secondary, yet unknown factor to fully express the hypertriglyceridemia phenotype. Severe hypertriglyceridemia also occurs secondary to other conditions including hypothyroidism and diabetes, however, usually not as severe. More importantly, in secondary hyperchylomicronemia the elevation in plasma VLDL remnant particles contributes to the hypertriglyceridemia phenotype. Patients are often diagnosed when the lactescent, grossly lipemic appearance of their blood raising the suspicion of chylomicronemia. The largest cohort of infants with familial chylomicronemia investigating presenting symptoms in these patients was done in Quebec [7]. 7/16 patients presented with irritability, 5 with anemia and/or splenomegaly, 2 had a rectal bleed and 2 cases were incidentally discovered. Another cohort of patients in South Africa of 29 individuals included adult patients. Here, the majority of patients presented with pancreatitis [8]. Patients who develop hypertriglyceridemia at a later age had less deleterious LPL mutations. If untreated, the massively increased TGs in the plasma leads to recurrent attacks of acute pancreatitis (the most common cause of early death in this population), hepatosplenomegaly, abdominal pain, eruptive xanthomata, and psychiatric manifestations [9] [10]. The only therapeutic intervention for patients with primary hyperchylomicronemia at present is a drastic reduction of fat in the patient's diet. For infants, milk feeds are substituted with MCT feeds. The fasting lipid levels in these patients respond usually within days to fat restriction. The mainstay of controlling the plasma concentration of lipids is dietary management, aiming at a daily intake of fat of less than 20 g equivalent to two glasses of whole milk. Compliance with this diet is difficult to maintain for the patient especially later in childhood and adolescence. In the young age, ensuring normal calorie intake from alternate sources to promote adequate growth is important, as is to ensure a balanced diet, especially in view of a restricted diet with vitamins, minerals and essential fatty acids. Supplementation with walnut oil is helpful as a source of essential fatty acids, compared to other edible oils. Exogenous factors such as alcohol, oral contraceptives, or retinoids may aggravate hyperlipidemia and should be avoided [9] [10]. Statins and other medications used in hyperlipidemias of other etiologies are not helpful and thus not indicated in patients with primary chylomicronemia. | ||||||
|
Conclusion
| ||||||
|
Chylomicronemia is rare, but its inclusion in the differential diagnosis of the unwell infant is important for the general pediatrician, especially because once recognized, the condition can be managed effectively by dietary intervention and potential serious complications can thus be avoided. Attempts to target monogenic chylomicronemia with gene therapy may alter the management approach in the future. | ||||||
|
Acknowledgement
| ||||||
|
We wish to thank Chris Smith, pediatric dietician at the Royal Alexandra Children's Hospital in Brighton, for critical revision of the manuscript. | ||||||
|
References
| ||||||
| ||||||

|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions
Susanna Felsenstein – Substantial contributions to conception and design, Acquisition of data, Drafting the article, Revising it critically for intellectually important content, Final approval of the version to be published Geesje Dallinga-Thie – Substantial contributions to analysis and interpretation of data, Revising it critically for important intellectual content, Final approval of the version to be published Shankar Kanumakala – Substantial contributions to analysis and interpretation of data, Revising it critically for important intellectual content, Final approval of the version to be published |
|
Guarantor of submission
The corresponding author is the guarantor of submission. |
|
Source of support
None |
|
Conflict of interest
Authors declare no conflict of interest. |
|
Copyright
© 2015 Susanna Felsenstein et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information. |
|
|