  |

|
 |
|
Case Report
| ||||||
| Miller–Dieker syndrome with hydronephrosis | ||||||
| Keya Lahiri1, Fehmida Najmuddin2, Rajesh Rai1, Priya Patil Cholera3 | ||||||
|
1Pediatrics, Professor, Department of Pediatrics, D Y Patil Medical College, Hospital & Research Centre, Maharashtra, India.
2Pediatrics, Assistant Professor, Department of Pediatrics, D Y Patil Medical College, Hospital & Research Centre, Maharashtra, India. 3Ophthalmology, Assistant Professor, Department of Ophthalmology, D.Y. Patil Medical College, Hospital & Research Centre, Maharashtra, India. | ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article]
[Similar article in Pumed] [Similar article in Google Scholar] 
|


| How to cite this article |
| Lahiri K, Najmuddin F, Rai R, Cholera PP. Miller–Dieker syndrome with hydronephrosis. Int J Case Rep Images 2015;6(10):610–613. |
|
Abstract
|
|
Introduction:
Miller–Dieker syndrome (MDS) is a rare genetic disorder which is characterized by lissencephaly, facial dysmorphism and congenital malformations involving-multiple organs.
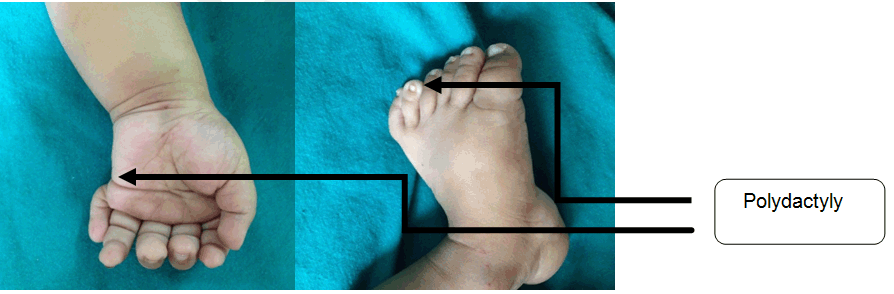
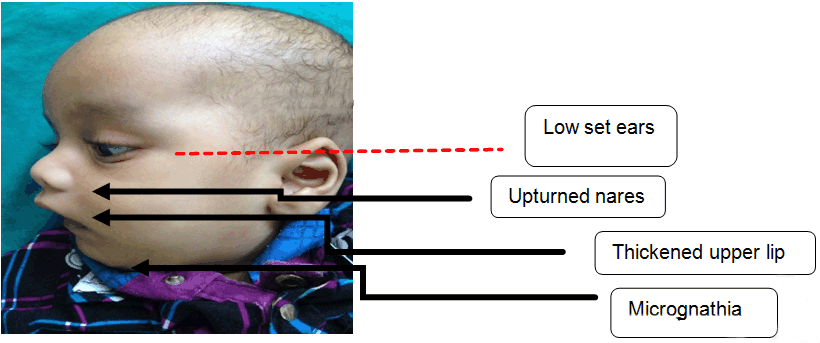
Case Report: We, hereby describe a nine- month-old infant who presented to our tertiary care hospital with developmental delay, infantile spasm and bronchopneumonia. On examination, there was prominent forehead, bi-temporal hallowing, bilateral ptosis, upturned nares and low set ears. Other dysmorphic features were micrognathia, thickened upper lip, high arched palate, umbilical hernia and polydactyly was noted bilaterally in both the upper and lower limbs. The investigations, revealed Lissencephaly type 1 and left-sided hydronephrosis. The typical dysmorphic facies, neurological involvement and lissencephaly type 1 led to the diagnosis of Miller–Dieker syndrome. Conclusion: Miller–Dieker syndrome involving the genitourinary system anomaly in the form of hydronephrosis with pelvi-ureteric junction obstruction, has not been yet described in literature. | |
|
Keywords:
Miller-dieker syndrome, Lissencephaly, Hydronephrosis
| |
|
Introduction
| ||||||
|
Miller–Dieker syndrome (MDS) results from a contiguous gene deletion involving 17p13.3 locus [1] . L1S1 gene is required for neurogenesis and neuronal migration and is located at 17p3. Miller–Dieker syndrome is usually associated with complete absence of this gene, which leads to classical Type I lissencephaly. Defect in the neuronal migration during embryonic development results in lissencephaly [2]. Apart from typical abnormal facies, MDS is also associated with intellectual disability, developmental delay, hypotonia and seizure disorder by first year of life. | ||||||
|
Case Report
| ||||||
|
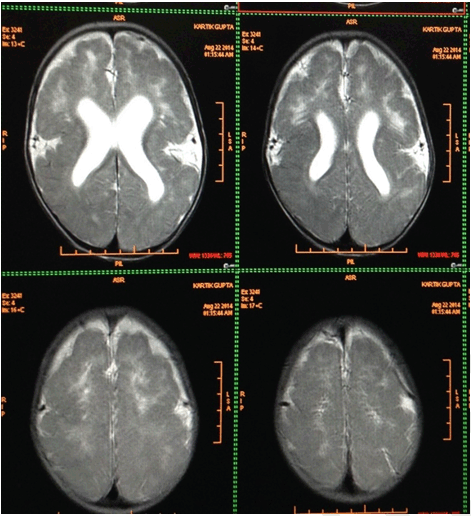
A nine-month-old male infant presented to a tertiary care hospital with complaints of inability to hold neck since five months of age, convulsions since three months, cough for a period of ten days and fever for two days. Convulsions were characterized by rapid forward bending of head and simultaneous movements of the arm suggestive of infantile spasms lasting for 2–3 seconds. Child was born of a non-consanguineous marriage with two well grown siblings. Mother did not attend the antenatal clinic. Infant was a full term normal vaginal delivery with birth weight of 3300 gms and was immunized till date. There was history of global developmental delay and the child belonged to a lower middle social class. On examination the patient was conscious, moderately built and nourished with a heart rate 112/min, respiratory rate 54/min, temperature 98.6°F and blood pressure 94/50 mmHg. Anthropometric measurements were normal for his age. On head to toe examination there was prominent forehead, bi-temporal hallowing, bilateral ptosis, upturned nares and low set ears. Polydactyly was noted bilaterally in both the upper and lower limbs (Figure 1). Other dysmorphic features revealed micrognathia, thickened upper lip, high arched palate and umbilical hernia (Figure 2). Cranial nerve involvement was in the form of bilateral ptosis with no response to sound and dysphagia with feeding difficulties. There was hypotonia with absent superficial reflexes and brisk deep tendon reflexes. There were no signs of meningeal irritation or cerebellar involvement. Other systems revealed bilateral crepitations along with hepatosplenomegaly. Hemoglobin was 8.9 g/dl, total leucocyte count 30,000/mm3 with neutrophils 64.3%, lymphocytes 30.2%, eosinophils 1.2%, monocytes 4.1% and platelets 6.01x105/mm3. The peripheral smear was suggestive of microcytic hypochromic anemia with neutrophilic leukocytosis and blood culture report was negative. Chest X-ray was suggestive of bronchopneumonia. Thyroid profile, serum electrolytes, calcium, alkaline phosphatase, liver and renal function tests were normal. Magnetic resonance imaging (MRI) scan of brain showed smoothening of the cortical surface of bilateral cerebral hemispheres with hypoplastic sulci and broad flattened gyri. The cerebral cortex was thickened, smooth well defined grey-white matter differentiation noted along with bilateral Sylvian fissures appearing shallow. All these features were suggestive of lissencephaly type I (Figure 3) Cranial ultrasonography did not reveal any abnormality. A computed tomography (CT) scan of abdomen and pelvis showed left sided gross hydronephrosis without hydroureter and marked thinning of cortex which was suggestive of pelvi-ureteric junction (PUJ) obstruction. An abnormal sleep EEG with right to left asymmetry and left occipital epileptogenesis was noted. Echocardiography and karyotyping were normal. The patient was administered injectable ceftriaxone (75 mg/kg/day) for 10 days. Syrup phenobarbitone (5 mg/kg/day) was started due to repeated episodes of convulsions. A surgical intervention for hydronephrosis has been planned. Classical dysmorphic features and type 1 lissencephaly characterized him as MDS. | ||||||
| ||||||
| ||||||
| ||||||
|
Discussion
| ||||||
|
Three types of lissencephaly have been described viz. Type-1 being the classical form which is associated with MDS. Type -2, also known as cobblestone lissencephaly is associated with o-glycosylation enzyme defect and type-3 is a neurodegenerative process with abnormal apoptosis. Apart from MDS, type-1 lissencephaly is also associated with Norman-Roberts syndrome, but these patients have severe microcephaly [3] . Apart from the characteristic dysmorphic features and lissencephaly, there was renal anomaly in the form of left-sided hydronephrosis with PUJ obstruction reported in our case. These children have an increased life expectancy due to better seizure control, nasogastric feeding and improved nutrition [4]. The most common cause of mortality associated with this syndrome is aspiration and recurrent respiratory infections [5]. A detailed antenatal history and regular antenatal visits is essential. History of polyhydramnios, intrauterine growth retardation and reduced fetal movements are associated with MDS. Prenatal ultrasonography findings in MDS are smooth gyral pattern, ventriculomegaly, large subarachnoid space, congenital heart disease and omphalocele [6] [7]. Classical lissencephaly can be detected on imaging only after 28 weeks of gestation. The recurrence risk for MDS is very low, as the chromosomal deletion is usually a de novo event. However, if it is associated with a familial reciprocal translocation, the recurrence risk for an abnormal live born can be as high as 33%. Normal results of fluorescent in situ hybridization (FISH) and chromosomal microarray tests cannot rule out partial and intragenic deletions or duplications of LIS1 gene [8]. The differential diagnosis of MDS includes Cornelia de Lange, Wolf-Hirschorn, Smith Lemli-Opitz and Zellweger syndrome as these syndromes too have facial dysmorphism, microcephaly, seizures and hypotonia but none of them are associated with lissencephaly [9] . The child has been gaining weight and has demonstrated reduced frequency of seizures on follow-up. | ||||||
|
Conclusion
| ||||||
|
To conclude, any child presenting with neurological involvement, lissencephaly and typical dysmorphism should be investigated for the above syndrome. | ||||||
|
References
| ||||||
| ||||||

|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions
Keya Lahiri – Substantial contribution to conception and design, Acquisition of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Fehmida Najmuddin – Substantial contribution to conception and design, Acquisition of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Rajesh Rai – Substantial contribution to conception and design, Acquisition of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published. Priya Patil Cholera – Substantial contribution to conception and design, Acquisition of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published |
|
Guarantor of submission
The corresponding author is the guarantor of submission. |
|
Source of support
None |
|
Conflict of interest
Authors declare no conflict of interest. |
|
Copyright
© 2015 Keya Lahiri et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information. |
|
|