 |

|
 |
|
Clinical Image
| ||||||
| Child with mucopolysaccharidosis type IV: Morquio syndrome | ||||||
| Ali Akhtar1, Sabina Manandhar2, Eswat Ahmad1 | ||||||
|
1Army Medical College, Rawalpindi, Punjab, Pakistan.
2Kathmandu University School of Medical Sciences, Nepal. | ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article]
[Similar article in Pumed] [Similar article in Google Scholar] 
|


| How to cite this article |
| Akhtar A, Manandhar S, Ahmad E. Child with mucopolysaccharidosis type IV: Morquio syndrome. Int J Case Rep Images 2015;6(10):658–660. |
|
Case Report
| ||||||
|
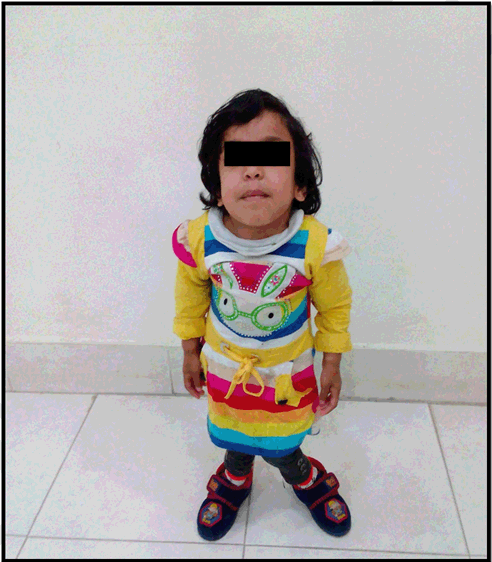
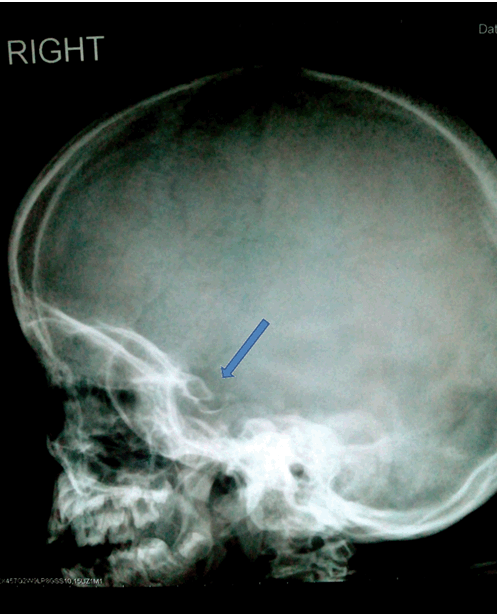
A five-year-old girl was presented at Armed Forces Institute of Radiology and Imaging for skeletal survey. On clinical examination she had corneal clouding, coarse facial features, short stature, and kyphotic deformity of spine. On radiography, skull was enlarged and J shaped sella, spine showed increased curvature and anterior beaking of vertebra. X-ray chest showed anterior widening of ribs (oar shaped ribs). Pelvis had widely flared iliac bones. Hand X-ray revealed proximal pointing of metacarpals of both hands. There was no mental retardation. She was diagnosed with mucopolysaccharidosis type IV: Morquio syndrome. | ||||||
| ||||||
|
| ||||||
| ||||||
|
Discussion
| ||||||
|
Mucopolysachharidoses (MPSs) are a family of rare, inherited (autosomal recessive) lysosomal storage disorders caused by deficiency of an enzyme involved in the degradation of glycosaminoglycans (GAGs): heparan sulfate, dermatan sulfate, keratan sulfate and chondroitin sulfate. They are classified as MPS I to MPSVII on the basis of clinical and biochemical studies [1]. Morquio and Brailsford reported cases of a disorder characterized by short neck, pectus carinatum, genu valga, pes planus, odontoid hypoplasia and normal intelligence independently. In 1960, this disorder was characterized as MPS caused by the lysosomal accumulation of GAGs and urinary excretion of the keratan sulfate [1]. Matalon et al. discovered MPS IVA as caused by deficiency of galactosamine-6-sulfate. Arbisser et al. described a patient with normal N-acetylgalactosamine-6-sulfate but deficient lysosomal b-galactosidase known later to be MPSIVB, the milder one [1]. Patients with MPS IVA appear normal at birth with normal intellect and experience clinical onset of disease during early childhood. The most common presentation is skeletal deformity and growth retardation in the second or third decade of life [2] [3]. The child in our case was also normal at birth with normal intellect. Other clinical features of Morquio syndrome include [4] [5] [6] [7]:
No dental abnormalities were noted in our case. In 2012, a familial tendency of unknown etiology has been described by Rekha et al. where three siblings in the same family were affected with the syndrome [8]. In 1952, Garn and Hurme described abnormalities of the teeth in nine siblings. Of them, three siblings showed thin enamel layer and in some places the dentine actually showed through. The surface was marked by numerous pits, and the enamel appeared to be structurally weak since it exhibited a tendency to fracture and flake off. In contrast, the six unaffected sibs had normal dentitions [9]. In our case, no other siblings were effected and history of consanguineous marriage was not elucidated. There are two treatment options for patients with MPS - Hematopoietic stem cell transplantation and recombinant intravenous enzyme replacement therapy. Early diagnosis and treatment can improve patient outcome and prolong survival [10]. This report outlines the clinical and radiological findings found in a case of Morquio syndrome. An accurate diagnosis typically requires the recognition of specific clinical and/or radiographic signs and symptoms together with laboratory confirmation. The radiologist can play a critical role in ensuring that an accurate diagnosis is reached expeditiously by raising suspicion of an MPS disorder if dysostotic multiplex changes are evident. | ||||||
|
Conclusion
| ||||||
|
Mucopolysaccharidosis (MPS) is a multisystem disorder and its diagnosis is based on clinical finding. Though it is a rare disease but its appropriate management and investigations are required to reach its diagnosis and treatment. Understanding the symptoms and progressive nature of MPS IV will provide a solid basis for evaluating the efficacy of treatment modalities. | ||||||
|
Keywords
| ||||||
|
Glycosaminoglycans, Joint hypermobility, Morquio syndrome, Mucopolysachharidoses, Visual impairment | ||||||
|
References
| ||||||
| ||||||

|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions
Ali Akhtar – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Sabina Manandhar – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Eswat Ahmad – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published |
|
Guarantor of submission
The corresponding author is the guarantor of submission. |
|
Source of support
None |
|
Conflict of interest
Authors declare no conflict of interest. |
|
Copyright
© 2015 Ali Akhtar et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information. |
|
|