| |
|
|
|
Case Report
| ||||||
| Macroglossia and periorbital ecchymoses in a patient with systemic amyloidosis: A case report | ||||||
| Jamille Hemétrio Salles Martins Costa1, Aloísio Benvindo de Paula2, Leonardo de Oliveira Campos3, Rafaela Brito de Paula4, Daniel Riani Gotardelo5 | ||||||
|
1Internal Medicine Resident, Hospital Márcio Cunha/FSFX; Ipatinga, MG, Brazil.
2Infectologist, Internal Medicine Residency Program Coordinator, Hospital Márcio Cunha/FSFX; Ipatinga, MG, Brazil. 3Neurologist, Hospital Márcio Cunha/FSFX; Ipatinga, MG, Brazil. 4Medical Student, Universidade Federal de Uberlândia; Uberlândia, MG, Brazil. 5Associate Professor, School of Medicine, Vale do Aço/Instituto Metropolitano de Ensino Superior; Ipatinga, MG, Brazil. | ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article]
[Similar article in Pumed] [Similar article in Google Scholar] 
|
| How to cite this article |
| Costa JHSM, de Paula AB, de Oliveira Campos L, de Paula RB, Gotardelo DR. Macroglossia and periorbital ecchymoses in a patient with systemic amyloidosis: A case report. Int J Case Rep Images 2015;6(6):343–347. |
|
Abstract
|
|
Introduction:
Amyloidoses comprise a group of rare diseases associated with the extracellular deposition of misfolded proteins, which can compromise the function of target organs and give rise to clinical disease with a broad range of manifestations. The aim of this study was to report a case of systemic amyloidosis with macroglossia and periorbital ecchymoses - two uncommon semiological findings.
Case Report: A 59-year-old female presented with dyspnea, vomiting, abdominal pain and distension. The patient was admitted for diagnostic workup, during which malnutrition, infiltrative thickening of the suprapubic abdominal wall, anasarca, macroglossia, and tongue petechiae were identified. The clinical picture was compounded by hematochezia and periorbital ecchymoses during hospitalization. Biopsy of the dermis and subcutaneous tissue of the hypogastrium revealed amorphous eosinophilic extracellular depositions on Congo red staining which had green birefringence under polarized light microscopy, consistent with amyloidosis. Conclusion: Patients with amyloidosis are usually extensively investigated before a diagnosis is made because in addition to being a rare disease with multifaceted presentation features, the signs and symptoms of amyloidosis are nonspecific. In the present report, cutaneous thickening with formation of periorbital ecchymoses accompanied by macroglossia were suggestive of amyloidosis, whose treatment and prognosis are influenced by timely diagnosis. | |
|
Keywords:
Amyloidosis, Macroglossia, Periorbital ecchymoses
| |
|
Introduction
| ||||||
|
Amyloidoses are a subgroup of diseases caused by the aggregation of misfolded proteins with extracellular deposition which compromises the function of target organs and gives rise to clinical disease. Amyloidosis is a rare disease and a diagnostic challenge because of its nonspecific presenting features [1] [2]. Being a rare disease, the exact incidence of amyloidosis is unknown. In the United States, incidence rates seem stable at around 6–10 cases/million/year. Older adults and males account 65–70% cases of the disease. The mean age at diagnosis is 64 years; Less than 5% patients are under 40 years of age at the time of diagnosis [3]. The term "amyloid" was attributed by Rudolph Virchow in 1854, when he noted a reaction of metachromasia to iodine in necropsied tissue samples, similarly to what occurs with starch, and assumed the material was of glycidic origin. Although Friedreich and Kekule demonstrated in 1859, that the material was in fact protein, the denomination was already incorporated into the medical vocabulary and was thus maintained. The "amyloid" deposit is necessarily composed of a fibrillar protein, glycosaminoglycans and serum amyloid P-component. Amyloid fibrils have a secondary structure in common-a beta-pleated sheet configuration-and a single ultrastructure which determines the 30 different precursor proteins known to date [4]. Amyloid diseases can be categorized as systemic or localized; hereditary or acquired. The current classification is based on the different types of protein of the amyloid fibrils, most often related to the distinct clinical presentations. The prognosis of localized disease is generally good with surgical treatment. If there is systemic involvement, the disease can be severe; with cardiomyopathy, nephrotic syndrome/renal failure, hepatosplenomegaly, diarrhea, intestinal pseudo-obstruction, peripheral neuropathy, autonomic neuropathy, arthropathy, carpal tunnel syndrome, bleeding, adrenal dysfunction, gout, weight loss, pulmonary problems, fatigue, and malaise [1]. A tissue biopsy and histopathological examination are done to establish the diagnosis. Amyloid deposition is identified using Congo red histological staining and subsequent observation of green birefringence under polarized light-the established gold standard. The precursor fibril is then characterized using histochemical and biochemical testing, and genetic analysis [5]. The correct and specific diagnosis of the amyloidosis type is essential to guide treatment. | ||||||
|
Case Report
| ||||||
|
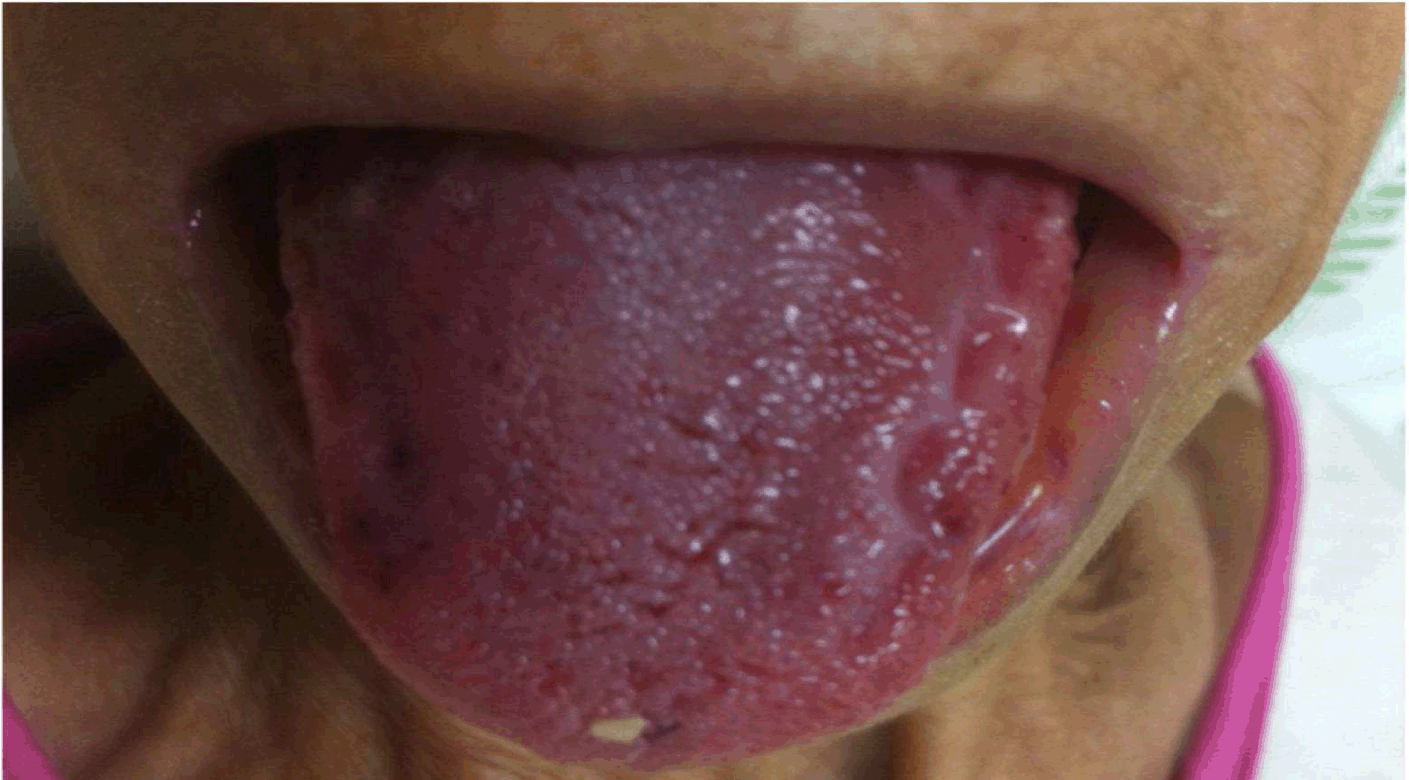
A previously healthy 59-year-old female was admitted with mild dyspnea, vomiting, abdominal pain and distension for diagnostic workup. The patient complained of lower abdominal heaviness in addition to postprandial bloating and decreased appetite of approximately one year duration, resulting in progressive weight loss that warranted extensive medical investigation at the time. She denied fever, inflammatory signs and changes in bowel habits. On physical examination, malnutrition, anasarca, macroglossia (Figure 1), tongue petechiae, and infiltrative thickening of the suprapubic abdominal wall were identified. During hospital stay, the disease progressed with hemorrhagic phenomenon consisting of massive hematochezia and bilateral periorbital ecchymoses in addition to extensive left-eye conjunctival hemorrhage (Figure 2). Pericardial effusion, bilateral pleural effusion and ascitis were noted on computed tomography scan. Abdominal magnetic resonance imaging showed edematous infiltration of the mesenterium and subcutaneous tissue in the hypogastrium. A barium meal revealed reduced small bowel motility. Hyperemia and ulcers were found in the colon and rectum on colonoscopy, the etiology to be determined by histopathology. Laboratory tests revealed antinuclear antibody (ANA), double-stranded deoxyribonucleic acid (ds DNA), rheumatoid factor (RF), hepatitis B surface antigen (HBsAg), anti-hepatitis C virus (Anti-HCV) antibody and human immunodeficiency virus (HIV) antigen within normal limit. Mantoux (with 10 U tuberculin) and alcohol acid-fast bacilli (sputum): were negatives. Blood glucose, urea, creatinine, sodium, potassium and bicarbonate levels were normal. Routine peripheral smear examination showed hypochromic microcytic anemia. C-reactive protein (CRP) 48 mg/L. TSH 6.06 ng/dL (0.27–4.2) ng/dL. free T4 1.19 ng/dL (0.93–1.7 ng/dL). Urinalysis was within normal limits except for a erythrocyturia (20/HPF). Fundoscopy showed abundant diffuse opacities in the vitreous. Electrophoresis of urine and blood proteins showed a monoclonal peak in the region of alpha-2-globulins. Immunohistochemistry of the bone marrow biopsy specimen was positive for CD138 with numerous plasma cells (90% of cell count), in addition to the presence of light-chain kappa and lambda globulins. Echocardiography showed grade III left ventricular diastolic dysfunction, pericardial effusion in addition to parietal and visceral thickening. Biopsy of a fragment of the dermis and subcutaneous tissue of the hypogastrium revealed amorphous eosinophilic extracellular deposits on Congo red staining and green birefringence under polarized light, both consistent with amyloidosis. The possibility of the disease being associated with multiple myeloma was excluded given the clinical manifestations and evidence from ancillary tests (absence of hypercalcemia, renal failure, or osteolytic lesions consistent with myeloma). Cardiac decompensation followed, as the patient was in a condition of severe advanced disease, while the medical team was expecting the results of the biopsy with Congo red staining. The patient died before the specific therapy was instituted. | ||||||
| ||||||
| ||||||
|
Discussion
| ||||||
|
Macroglossia is the most frequent oral manifestation of amyloidosis and can be found as the only presenting symptom or as only one of the symptoms of the disease. Before considering the presence of amyloid protein, other more likely causes of tongue enlargement, such as malignant tumors of the tongue, vascular abnormalities, hypothyroidism and deficiency of vitamin B12 and folic acid, should be considered. Xavier et al. described a case of an older adult with macroglossia, weight loss, and dysphagia that seemed at first to be a malignant tumor of the tongue and that, after proper workup, was defined as amyloidosis [6]. Tsourdi et al. reported a case of macroglossia as the sole manifestation of amyloidosis secondary to a monoclonal gammopathy of undetermined significance [7]. Purpuric eruptions such as ecchymoses and hematomas can also be found in individuals with amyloidosis. They are due to coagulation factor X deficiency, likely the result of its absorption by the amyloid fibrils, in addition to amyloid infiltration of capillaries causing microvascular fragility. Few cases have been described in literature in which the two findings-macroglossia and periorbital ecchymoses-were concurrent in patients with systemic amyloidosis; in most such cases, amyloidosis was associated with multiple myeloma [8][9]. In the case reported herein, no underlying diseases were found to account for the amyloid deposition, hence the diagnosis of primary systemic amyloidosis was considered. This type of amyloidosis is known as AL, with the first letter ("A") corresponding to "amyloidosis" and the second representing the biochemical makeup of the constituent fibril-in this case, amyloidosis involving the deposition of light-chain immunoglobulins ("L" for "light-chain"). Three of the four diagnostic criteria to confirm AL-type systemic amyloidosis were verified in our study: (1) presence of a syndrome related to the amyloid deposits (heart failure and macroglossia, among other signs and symptoms); (2) evidence of amyloid deposition on Congo red staining in a tissue biopsy sample; and (3) presence of monoclonal plasma cell proliferation. The fourth diagnostic criterion would be the confirmation of light-chain proteins in the amyloid material through immunohistochemistry or other molecular biology techniques. These tests were not performed because of the rapidly fatal outcome [10]. The prognosis of AL amyloidoses is typically poor. Heart failure and renal failure are the main causes of death. When amyloidosis is secondary to multiple myeloma, the mean survival is five months, while the primary form of the disease is associated with a survival of 2.1 years. The treatment for AL amyloidosis is intended to reduce the amount of circulating precursor proteins produced by B-lymphocytes and plasma cells, which can be achieved with cytotoxic agents such as prednisone and melphalan [1]. | ||||||
|
Conclusion
| ||||||
|
Macroglossia, periorbital ecchymoses, and other hemorrhagic manifestations are among the multiple presenting features to be found in systemic amyloidosis, which is a severe disease with complex symptomatology requiring thorough clinical examination and early recognition by the medical team to ensure timely treatment. | ||||||
|
References
| ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions
Jamille Hemétrio Salles Martins Costa – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Aloísio Benvindo de Paula – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Leonardo de Oliveira Campos – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Rafaela Brito de Paula – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Daniel Riani Gotardelo – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published |
|
Guarantor of submission
The corresponding author is the guarantor of submission. |
|
Source of support
None |
|
Conflict of interest
Authors declare no conflict of interest. |
|
Copyright
© 2015 Jamille Hemétrio Salles Martins Costa et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information. |
|
|

|
Jamille Hemétrio Salles Martins Costa is Internal Medicine Resident, Hospital Márcio Cunha/FSFX; Ipatinga, MG, Brazil.
|
|
|
Aloísio Benvindo de Paula is Infectologist, Internal Medicine Residency Program Coordinator, Hospital Márcio Cunha/FSFX; Ipatinga, MG, Brazil.
|
|
|
Leonardo de Oliveira Campos is Neurologist, Hospital Márcio Cunha/FSFX; Ipatinga, MG, Brazil.
|
|
|
Rafaela Brito de Paula is Medical Student, Universidade Federal de Uberlândia; Uberlândia, MG, Brazil
|
|
|
Daniel Riani Gotardelo is Associate Professor, School of Medicine, Vale do Aço/Instituto Metropolitano de Ensino Superior; Ipatinga, MG, Brazil.
|