  |

|
 |
|
Case Report
| ||||||
| Idiopathic pulmonary hemosiderosis without hemoptysis in an adult: A rare presentation | ||||||
| Jackin Moses R1, Nishant Sinha1, Madhusmita M1, Kisku K H1, Manjiri P1 | ||||||
|
1Pondicherry Institute of Medical Sciences, Kalapet, Pondicherry, 605014.
| ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article]
[Similar article in Pumed] [Similar article in Google Scholar] 
|


| How to cite this article: |
| Jackin MR, Singha N, Madhusmita M, Kisku KH, Manjiri P. Idiopathic pulmonary hemosiderosis without hemoptysis in an adult: A rare presentation. International Journal of Case Reports and Images 2014;5(3):230–234. |
|
Abstract
|
|
Introduction:
Idiopathic pulmonary hemosiderosis (IPH) is defined as a clinical triad of hemoptysis, pulmonary infiltrates, and iron deficiency anemia. It is a diagnosis of exclusion when all other causes of diffuse alveolar hemorrhage have been ruled out. Epidemiologically, IPH is a rare disease, with an incidence of 0.24 per million children per year; overall, 80% of cases occur in children, most being diagnosed in the first decade of life.
Case Report: We present a 38-year-old female who presented to our casualty with acute respiratory failure which resolved on high dose corticosteroids. Patient on evaluation was diagnosed to have diffuse alveolar hemorrhage with idiopathic pulmonary hemosiderosis. Patient is currently asymptomatic and is on regular follow-up in our hospital. Conclusion: Diffuse alveolar hemorrhage should be a differential diagnosis in a patient presenting with hemoptysis, breathlessness, anemia and radiological evidence of infiltrates. Awaiting detailed work-up, patients should be promptly started on high dose steroids. | |
|
Keywords:
Idiopathic pulmonary hemosiderosis (IPH), Diffuse alveolar hemorrhage (DAH), Acute interstitial pneumonia
| |
|
Introduction
| ||||||
|
Idiopathic pulmonary hemosiderosis (IPH) is defined as clinical triad of hemoptysis, pulmonary infiltrates, and iron deficiency anemia. [1] [2] It is a diagnosis of exclusion when all other causes of diffuse alveolar hemorrhage have been ruled out. [3] [4] Epidemiologically, IPH is a rare disease, with an incidence of 0.24 per million children per year; [5] overall, 80% of cases occur in children, most being diagnosed in the first decade of life. [6] The remaining 20% of cases are adult-onset IPH, most of which are diagnosed before 30 years of age. In adults it is more common in males and in smokers. [6] Our patient had a rare presentation as she is a 38-year-old female, non smoker and had no history of hemoptysis. | ||||||
|
Case Report
| ||||||
|
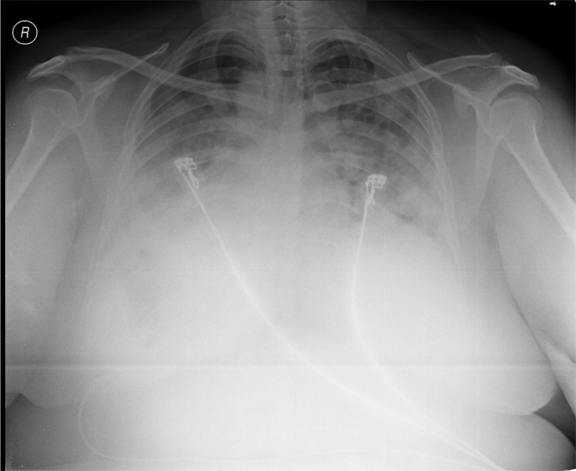
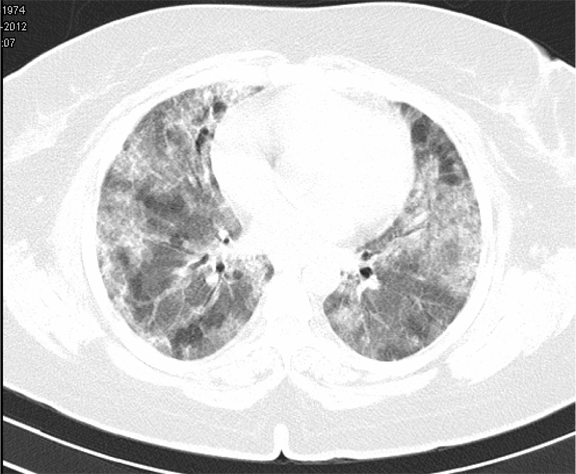
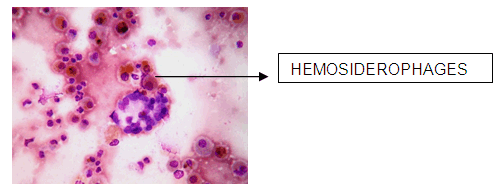
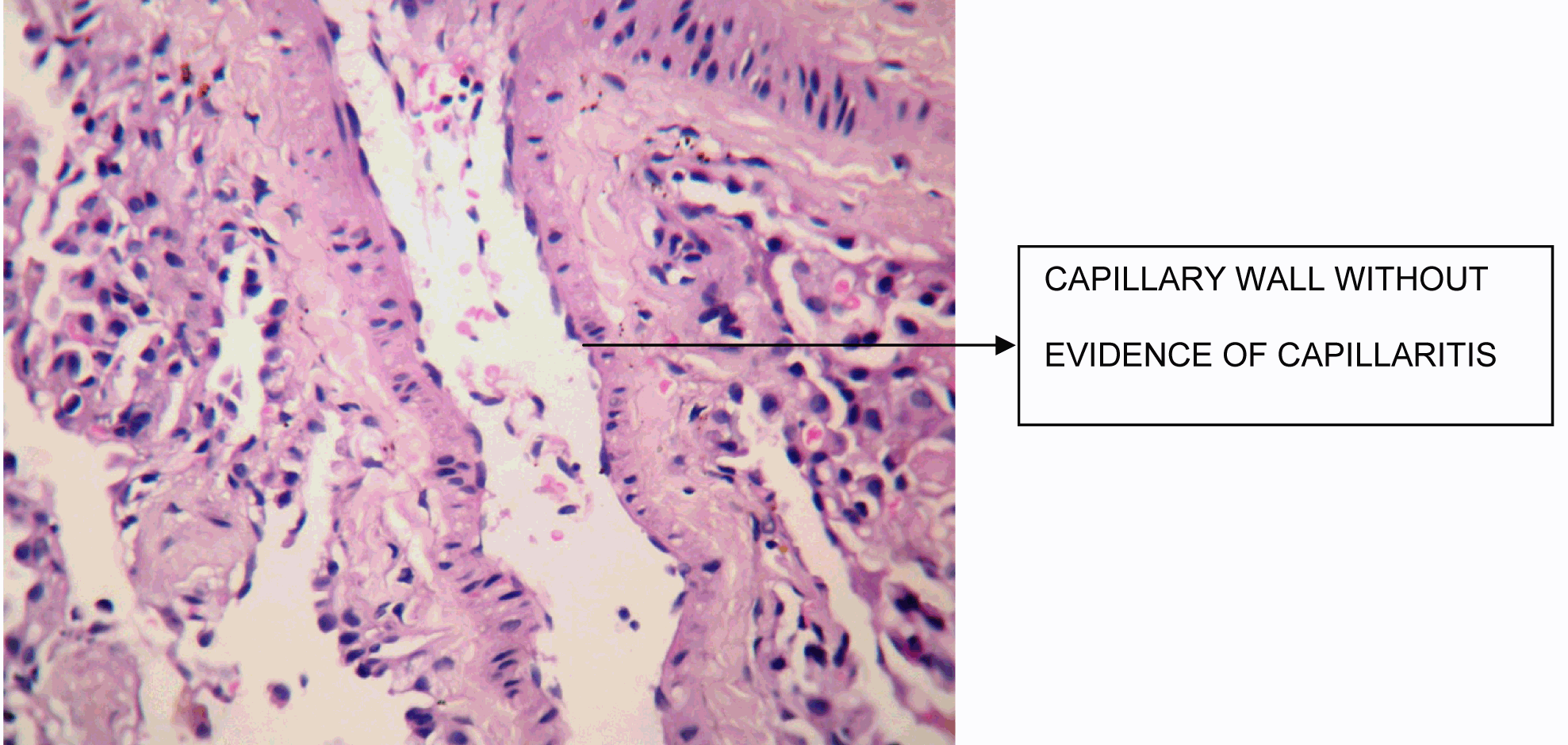
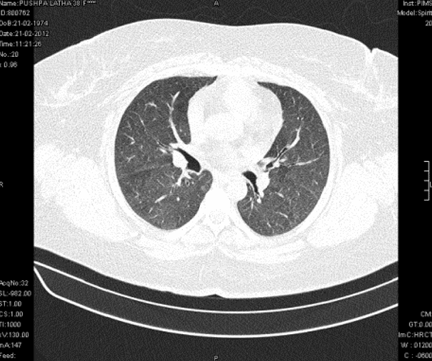
A 38-year-old female, presented to our emergency room with progressive breathlessness (MMRC grade 2- 4) and dry cough for three months. She had history of multiple blood transfusions elsewhere during the prior three months period. She had no history of hemoptysis, hematemesis, menorrhagia, bleeding per rectum or any other obvious focus of bleeding. There was no history of fever. She had no symptoms related to connective tissue disorder. She was not a known smoker and had no history of exposure to toxic gases and fumes. She had no history of exposure to any drugs. Physical examination in the emergency room revealed the temperature to be 370 °C, pulse rate 130 beats/minute, Blood pressure 130/80 mmHg, Respiratory rate 60/minute and SpO2 was 56% on room air ventilation. Her palpebral conjunctiva was pale. On auscultation fine end inspiratory crackles were heard bilaterally in all lung fields. Physical examinations of other systems were found in normal limits. Arterial blood gas analysis of the patient showed Ph 7.46, PCo2 30 mmHg, Po2 37 mmHg, SpO2 77 mmHg (P/F ratio of 0.37). Her serum d-dimer done within two hours of admission was low. Electrocardiography and echocardiogram were normal. Her total counts were also normal. She was admitted in intensive care unit (ICU) and was treated with non invasive ventilation and high flow oxygen. Subsequent investigations revealed corrected reticulocyte count 0.5%, microcytic hypochromic peripheral blood smear, low serum iron (36 μg/dL), low transferrin saturation (28%), normal Ferritin (140.2 ng/mL), normal total iron binding capacity (415 μg/dL) and normal serum vitamin B12 and folic acid levels. Her stool was negative for occult blood or parasites. Her serum creatinine was 0.8 mg/dL, blood urea was 36 mg/dL and urine microscopy was normal (no protein or red blood cells). The liver function tests and coagulation profile were within normal limits. Her retroviral status (HIV I and II by ELISA), hepatitis B surface antigen and hepatitis C antibody were negative. She received one pint blood transfusion in the ICU. Her chest X-ray (Figure 1) which was done in the ICU showed bilateral reticular infiltrates. Computed tomography (CT) scan of thorax was subsequently done which showed bilateral extensive ground glass opacities with interstitial thickening and nodules. (Figure 2) A provisional diagnosis of acute interstitial pneumonia/diffuse alveolar hemorrhage was made and she was started on broad spectrum antibiotics with high dose pulse steroids (Inj methylprednisolone 500 mg i.v. daily in divided doses for 3 days). Within 3 days she improved significantly and was weaned off the non invasive ventilator support. Later the steroids were tapered to oral prednisolone at 0.75 mg/kg body weight. Subsequently fibre optic bronchoscopy was done under local anesthesia with conscious sedation. Sequential broncho alveolar lavage (BAL) was done from right middle lobe, it showed increasing hemorrhagic returns of BAL aliquots and BAL counts showed 60% hemosiderophages, (Figure 3) confirming the diagnosis of diffuse alveolar hemorrhage. BAL was negative for gram stain and culture showing no evidence bacterial pathogens, acid fast bacilli, fungal organism, Fluorescent antibody stain for Pneumocystis jeroveci and cytology for cancer cells. Transbronchial lung biopsy was done from right middle and lower lobes. It showed interstitial fibrosis, hemosiderin laden macrophages in alveolar spaces and interstitium with positive Perl’s stain. (Figure 4) There was no evidence of vasculitis (capillaritis), granulomas, organising pneumonia and malignancy in the biopsy specimen. Her serum Rheumatoid factor (RF), antinuclear antibody (ANA), Double stranded Deoxyribonucleic acid (DsDNA), antiphospholipid antibody(APLA), Anti Glomerular Basement Membrane antibody (anti GBM) and anti U1 RNP were negative. Serum Complements (C3, C4 and Total Complement) and Antineutrophilic Cytoplasmic antibodies(c and p ANCA) were in normal range. After one week of treatment with steroids, she improved and there was no further drop in hemoglobin. At this time spirometry was done and it was normal. Her chest X-ray and CT thorax repeated after one month showed complete clearing of the shadows. (Figure 5) | ||||||
| ||||||
| ||||||
| ||||||
| ||||||
| ||||||
|
Discussion
| ||||||
|
The entity of IPH was initially described by Virchow [7] in 1864 as “brown lung induration”. Waldenstrom [8] established the first ante mortem diagnosis in 1944. There are a few aetiological hypotheses of IPH including genetic, autoimmune, allergic, environmental and metabolic theory. [9] Environmental exposure to fungi (especially Stachybotrys atra) in water-damaged houses has been mentioned to cause IPH. [10] The clinical course includes two phases. First, an acute phase “IPH exacerbation”, corresponds to intra-alveolar bleeding episodes, manifested by cough, dyspnea, hemoptysis and sometimes respiratory failure. Almost 100% of the adults experience hemoptysis during the disease course. [9] Secondly, the chronic phase is characterised by a slow resolution of previous symptoms, with or without treatment. The alternation of the two phases is largely unpredictable. Our patient never had hemoptysis. The diagnosis of IPH is difficult, it requires the exclusion of other diseases which can be associated, such as cardiac diseases, bleeding disorders, connective-tissue diseases (systemic lupus erythematosus and rheumatoid arthritis), systemic vasculitis (Wegner’s granulomatosis or microscopic polyangiitis), or anti-basement-membrane-antibody diseases (Goodpasture’s syndrome). Laboratory analysis for ANCA, anti-GBM, antiphospholipid antibody, ANA and RF should so be negative. In our patient, they were all negative. Association of IPH with celiac disease has been mentioned in literature. [11] However, we have not done serum antigliadin, antireticulin and cow-milk precipitins as she had no symptoms suggestive of celiac disease or milk allergy. Goodpasture’s syndrome can present with or without pulmonary capillaritis, [3] diffuse alveolar hemorrhage resulting from antibasement membrane antibody disease is usually associated with glomerulonephritis. [1] Immunofluorescence of transbronchial lung biopsy specimen was not done as she had no glomerulonephritis and serum anti GBM antibody was negative. Based on the clinico- radio- pathological interpretation, we feel that the findings are consistent with idiopathic pulmonary hemosiderosis. A number of therapeutic trials have been tried, including: (1) splenectomy, without significant results (there is no evidence of hypersplenism), and (2) systemic glucocorticoid, commonly started in the acute phase of IPH, with apparent good control and impact on mortality. [1] Among immunosuppressants, azathioprine in combination corticosteroids might be the best therapeutic regimen, especially in preventing IPH exacerbations. [12] Adults with IPH seem to respond more favorably to oral corticosteroid therapy [6] than children, in whom treatment can be problematic. The recommended starting dose is 1 mg/kg/day prednisolone for two months, until the new alveolar infiltrates tend to resolve. [13] [14] In our patient, there was complete radiological clearing at four weeks and repeat CT thorax after four weeks showed no new infiltrates. We treated our patient with oral prednisolone 0.75 mg/kg body weight for 4 weeks, followed by 0.5 mg/kg body weight for another 8 weeks and 0.25 mg/kg body weight for another 12 weeks (Total duration of 6 months). We had planned to add azathioprine if she had recurrence or exacerbation. She is in our regular follow-up and has no recurrence of her symptoms. The long-term prognosis of the disease remains poor, and death may be due to massive diffuse pulmonary hemorrhage or chronic respiratory failure because of pulmonary fibrosis. [11] | ||||||
|
Conclusion
| ||||||
|
Diffuse alveolar haemorrhage should be a differential diagnosis in a patient presenting with hemoptysis, breathlessness, iron deficiency anemia and radiological evidence of infiltrates. Awaiting detailed work up, patients should be promptly started on high dose steroids. Idiopathic pulmonary hemosiderosis is confirmed when all other secondary causes of alveolar hemorrhage have been ruled out. Adults with IPH respond better to steroid therapy than children. | ||||||
|
References
| ||||||
| ||||||

|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions
Jackin Moses R – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Nishant Sinha – Substantial contributions to conception and design, Drafting the article, Final approval of the version to be published Madhusmita M – Substantial contributions to conception and design, Revising it critically for important intellectual content, Final approval of the version to be published Kisku KH – Substantial contributions to conception and design, Revising it critically for important intellectual content, Final approval of the version to be published Manjiri P – Substantial contributions to conception and design, Revising it critically for important intellectual content, Final approval of the version to be published |
|
Guarantor of submission
The corresponding author is the guarantor of submission. |
|
Source of support
None |
|
Conflict of interest
Authors declare no conflict of interest. |
|
Copyright
© Jackin Moses R et al. 2014; This article is distributed the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any means provided the original authors and original publisher are properly credited. (Please see Copyright Policy for more information.) |
|
|