| Table of Contents |  |
|
Case Report
|
| Cardiac transplantation for dilated cardiomyopathy in a patient with friedreich's ataxia: A case report |
| Federico A Silva Sieger1, Gustavo A Díaz Silva2, Mario A Ardila Vera2, María F Saavedra Chacón2, Sylvia C Méndez Díaz3 |
|
1Neurovascular science group chief, Neurosciences Department, Fundación Cardiovascular de Colombia (FCV), Floridablanca, Santander, Colombia.
2Neuroscience research fellowship, Neurosciences Department, Fundación Cardiovascular de Colombia (FCV), Floridablanca, Santander, Colombia. 3PM & R, Neurosciences Department, Fundación Cardiovascular de Colombia (FCV), Floridablanca, Santander, Colombia. |
|
doi:10.5348/ijcri-2012-06-134-CR-7
|
|
Address correspondence to: Federico Arturo Silva Sieger Calle 155a # 23-58, 3er piso Fundación Cardiovascular de Colombia Floridablanca, Santander Colombia Phone: 577- 6399292 Ext. 255 Fax: 577-6392744 Email: federicosilva@fcv.org |
|
[HTML Abstract]
[PDF Full Text]
|
| How to cite this article: |
| Silva Sieger FA, Díaz Silva GA, Ardila Vera MA, Saavedra Chacon MF, Mendez Díaz SC. Cardiac transplantation for dilated cardiomyopathy in a patient with friedreich's ataxia: A case report. International Journal of Case Reports and Images 2012;3(6):30–33. |
|
Abstract
|
|
Introduction:
Friedreich's Ataxia (FA) is a hereditary spinocerebellar degenerative disease, whose main features include ataxia, dysarthria and lower limb arreflexia. Cardiomyopathy is an important cause of mortality in these patients and usually a late finding.
Case Report: We present a case of a young adult who underwent cardiac transplantation for cardiomyopathy of unknown cause in terminal state. This patient attended neurology consult six months after transplant due to gait instability, balance disturbances and dysarthria. The diagnosis of Friedreich's Ataxia was confirmed by genetic testing. Conclusion: Few cases of Friedreich's Ataxia presenting as early cardiomyopathy have been published in the literature and to our knowledge this is the third report of cardiac transplantation in patients with this neurologic condition. This is probably because in most cases neurological disease is well established when cardiac compromise is evident, precluding the transplantation. Cardiac transplantation in these patients may improve life quality in the short term, but doesn't modify the disease's natural progression. | |
|
Key Words:
Cardiac transplantation, Friedreich's ataxia, Dilated cardiomyopathy, Trinucleotide repeats
| |
|
Introduction
| ||||||
|
Friedreich's Ataxia (FA) is a spinocerebellar degenerative disease, with an autosomal recessive inherited pattern, genotypically characterized by hyperexpansion of GAA triplet in the first intron of the frataxin gene in the chromosome 9. [1] Ataxia, dysarthria, lower limb arreflexia and extensor plantar responses are the most commonly found features. Cardiac compromise comprises a large fraction of mortality in these patients. [2] In most cases of cardiac involvement secondary to FA, the cardiomyopathy is a late feature while the early cardiomyopathy is an unusual finding. [3] Only five cases of early cardiomyopathy associated to FA have been published in the literature and to our knowledge this is the third report of cardiac transplantation in patients with this neurologic condition. [4] We present the case of a 22-year-old patient with dilated cardiomyopathy of unknown cause in terminal state that required cardiac transplantation, presenting six months later with gait, balance and language disturbances, leading to a presumptive diagnosis of FA, confirmed by genetic testing. | ||||||
|
Case Report
| ||||||
|
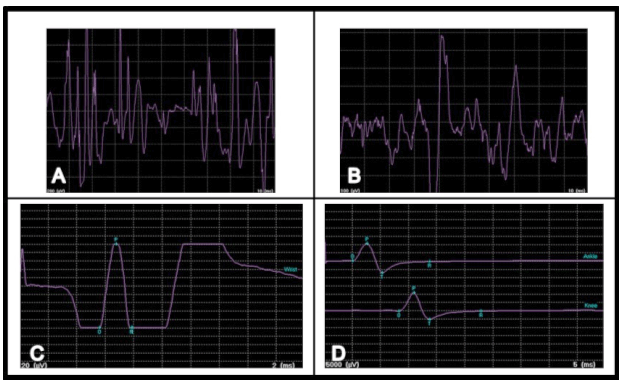
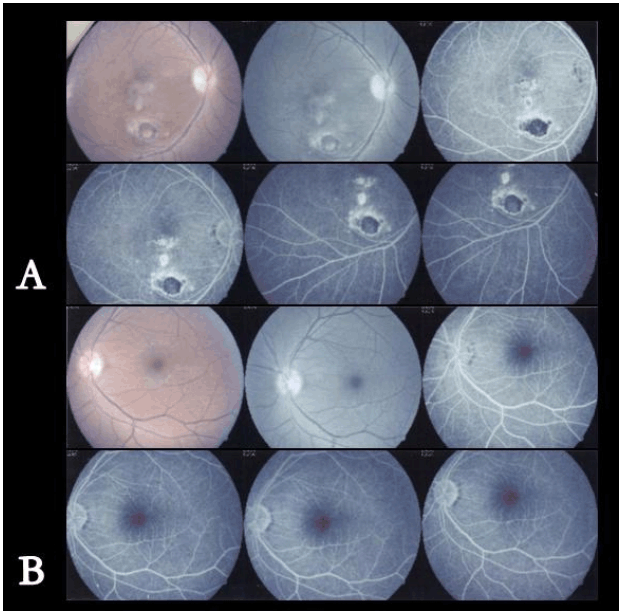
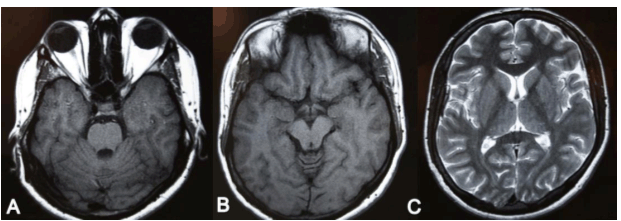
A 22-year-old male patient diagnosed with congestive cardiac failure since age of 18 years presented to emergency service with worsening of the functional class, progressive dyspnea, orthopnea and syncopal episodes. Physical examination revealed jaundice, hypotension, tachycardia, jugular venous distension, hepatojugular reflux sign, rhythmic heart sounds with a mitral murmur, rales in pulmonary bases and painful hepatomegaly. X-ray showed an enlarged cardiac silhouette and bilateral reticulonodulary opacities. Electrocardiography showed signs of dilation of the cardiac cavities. Echocardiographic findings included a severely dilated and hypokinetic left ventricle (ejection fraction less than 10%) and plurivalvular insufficiency without any structural valve damage. Cardiac catheterization confirmed these findings and showed no coronary lesions. Tests for Chaga's disease as well as other infectious and metabolic markers were negative. The patient underwent an uncomplicated cardiac transplantation after three months on waiting list, with an uneventful post-transplantation recovery. During cardiology and cardiovascular surgery follow-up he showed an excellent systolic function (ejection fraction more than 60%), but after six months he complained about gait instability that limited his walking, so he was referred to neurology consultation. After careful history taking the patient acknowledged that since age 15 he had noted a mild lower limb weakness that caused a barely perceptible disturbance in his gait pattern and that in the following years it progressed to gait instability, ataxia, dysmetria and dysarthria. These symptoms had stabilized after transplantation but subsequently worsened in the following months. Upon neurological examination important gait and stance ataxia, dysarthria, lower limb arreflexia and extensor plantar responses were found, as well as pes cavus, hammer toes and mild kyphoscoliosis. This all lead to a clinical diagnosis of FA, confirmed after genetic testing which revealed a homozygous hyper-expansion of 900 and 700 triplets in the frataxin gene. In addition he was diagnosed with Diabetes Mellitus. The patient was lost from neurology consult for the following eight years, during which his ataxia and language disturbance worsened, in spite of keeping an adequate cardiac function. He underwent nerve conduction studies and electromyography, whose results suggested a severe neuropathic pattern (Figure 1C, D). Visual evoked potentials showed disturbances of post-quiasmal visual pathway and the fluorescein angiography showed bilateral optic neuropathy and pigmentary retinopathy in the right eye (Figure 2). Cerebellar atrophy was detected by magnetic resonance imaging (MRI) scans (Figure 3). Eight years later, the patient's neurologic function is severely affected, with decreasing visual acuity, dysarthria, glove and stocking hypoesthesia, arreflexia and marked limb hypotrophy and ataxia that impairs his deambulation, confining him to a wheelchair. | ||||||
|
| ||||||
| ||||||
| ||||||
|
Discussion
| ||||||
|
Friedreich ataxia is an autosomal recessive spinocerebellar degenerative disorder described for the first time by Nicholaus Friedreich in 1863. This disorder is actually recognized as the most common of the inherited ataxias, with an estimate frequency of presentation in general population around 1 case per 29000 to 50000 habitants. [1] [2] It's genotypically characterized by a triplet GAA repeat expansion in the first intron of the frataxin gene on chromosome 9, which results in the loss of function of this mitochondrial matrix protein involved in iron metabolism and regulation, as well as the cellular respiratory chain. [1] [5] The expansion length varies from 7 to 29 triplets in normal subjects; however the patients with FA often present 66 to 1360 triplets. [4] The presented case was determined by a homozygous GAA expansion of 700 and 900 repeated triplets in each chromosome. Usually this disorder becomes evident during puberty, although singular cases with onset from 2 to 25 years old have been reported. [2] [4] The most common clinical manifestations are limb and postural ataxia, muscle weakness, dysarthria, dysmetria, volitional tremor, loss of vibration sense and position, lower limbs arreflexia, plantar extensor response and distal amyotrophy, often associated with osteomuscular deformities such as scoliosis, pes cavus and hammer toes. [2] [4] Approximately 25% of subjects with FA develop optic disorders, mainly accounted by fixation instability, optic atrophy and nystagmus, whereas 20% have hypoacusia. [6] As the disease advances dysphagia develops, which can be severe. Neurophysiologic studies often show more reduced sensorium than motor conduction velocities. [7] Histopathological features include loss of large myelin fibers from the dorsal ganglia neurons with secondary axonal degeneration of the spinocerebellar and corticospinal tracts at the latest stage of the disease. [7] Additionally, peripheral nerve sensory fiber involvement is observed. Most of these clinical and neurophysiologic manifestations were observed in our patient. Despite preponderant neurologic compromise, this disease is also related with cardiac, pancreatic and osteomuscular disorders, due to the high content of mitochondria in these tissues. Since the first descriptions of this syndrome by Friedreich, the cardiac compromise has been highlighted. Subsequent descriptions made by Pitt et al. [8] and Mollaret et al., [9] Sayuri et al. [10] has left to consider the cardiac compromise as one of the most classic features of this disorder, found in up to 96% of affected subjects. Cardiac histopathological findings in FA subjects include cardiomyocyte hypertrophy and lipid degeneration with interstitial fibrosis and infiltrating lymphocytes and eosinophils. [7] The cardiac compromise is recognized as the main cause of death among FA patients. However, the cardiac manifestations are unusual in the early course of the disease. Only five cases of early FA related cardiomyopathy have been reported. [3] [4] In our case, the patient underwent multiple studies in search of the cause of cardiomyopathy, however none of the tests showed alterations, including the Chaga's test. Hypertrophic cardiomyopathy and conduction system anomalies are the most frequent manifestations reported in FA, while dilated cardiomyopathy and coronary disease are unusual findings. Usually the cardiac compromise presents a rapid progression, finally developing lethal cardiac failure. [3] [4] [11] To the best of our knowledge, only two cases of cardiac transplantation for this condition have been reported. This is probably because of the poor neurological prognosis by the time of cardiomyopathy diagnosis. [4] In our case, the patient with FA developed a severe dilated cardiomyopathy, this case being the third published case of cardiac transplantation in a FA patient. The first case of cardiac transplantation in a patient with FA was described in a four month pediatric patient with an "idiopathic" cardiomyopathy, who developed neurologic symptoms after the procedure. [12] The other case differed in that it was a previously diagnosed FA patient, in whom cardiac compromise was so severe that no other option was available but the transplantation. [13] Surprisingly both cases reported some short term improvement of their neurologic function and better life quality in spite of progressive neurodegeneration. The second case even described a subjective improvement in ataxia symptoms. It's unclear if the natural history and final prognosis of FA is modified by cardiac transplantation. We have been able to perform a longer follow up than the other reports. In our patient ataxia continued its ruthless progression, impairing deambulation and confining him to a wheelchair, even though some improvement has been seen in symptoms like dysarthria and dysmetria, perhaps in association with language and occupational therapy. However, during this time, the patient has had a good performance in his familiar core. Moreover, his neuro-psychological evaluation was normal. Although in the aforementioned cases some life quality improvement was noticed in short-term follow up, this subjective effect may be accounted by cardiac function improvement and a true effect of cardiac transplantation in neurologic progression can't be acknowledged. In the presented case, the patient has had eight years of survival and his actual cardiac function is normal. In these cases it's important to note that the average life of cardiac transplantation is around nine years and re-transplantation is technically very difficult to perform and carries a worse prognosis and high mortality rate. | ||||||
|
Conclusion
| ||||||
|
Cardiac transplantation for dilated cardiomyopathy due to Friedreich ataxia may improve functional classification and life quality in the short term, but doesn't seem to modify the natural history and progression of the disease. In the absence of conclusive evidence, the role of cardiac transplantation in these patients remains unclear. We believe each case should be individualized, according to the degree of neurologic compromise and its impact before making a therapeutic decision. | ||||||
|
Acknowledgements
| ||||||
|
This study was supported by Colciencias, project code # 656651929173. | ||||||
|
References
| ||||||
| ||||||

|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions:
Federico Silva Sieger - Conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Critical revision of the article, Final approval of the version to be published Gustavo A Díaz Silva - Conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Critical revision of the article, Final approval of the version to be published Mario A Ardila Vera - Conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Critical revision of the article, Final approval of the version to be published María F Saavedra Chacón - Conception and design, Acquisition of data, Analysis and nterpretation of data, Drafting the article, Critical revision of the article, Final approval of the version to be published Sylvia C Méndez Díaz - Conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Critical revision of the article, Final approval of the version to be published |
|
Guarantor of submission:
The corresponding author is the guarantor of submission. |
|
Source of support:
None |
|
Conflict of interest:
Authors declare no conflict of interest. |
|
Copyright:
© Federico A Silva Sieger et al. 2012; This article is distributed the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any means provided the original authors and original publisher are properly credited. (Please see Copyright Policy for more information.) |
|
|