| Table of Contents |  |
|
Case Report
|
| Mohr syndrome: A rare case of oro-facial-digital syndrome type II with congenital heart disease |
| Prakashgouda HK Goudar1, Ravindra Joshi2, Shivaprakash V Hiremath3, Pramod Bhimrao Gai4 |
|
1Junior Research Fellow Research Centre for DNA Diagnostics, Department of Applied Genetics Karnatak University, Dharwad, Karnataka, India.
2Senior Consultant Pediatrician, Chetana Children Clinic Narayanpur Road-Dharwad, Karnataka, India. 3Associate Professor & Head of Department, Department of Biotechnology, K. L. E. Society, P. C. Jabin Science College, Vidyanagar. 4Professor and Co-ordinator, Department of Applied Genetics, Principal Investigator, Research Centre for DNA Diagnostics, Karnatak University, Dharwad, Karnataka, India. |
|
doi:10.5348/ijcri-2012-02-94-CR-8
|
|
Address correspondence to: Dr. Pramod Bhimrao Gai Professor and Co-ordinator, Department of Applied Genetics, Principal Investigator Research Centre for DNA Diagnostics, Karnatak University Dharwad - 580003, Karnataka India Phone: 0836- 2446274 Fax: 0836-2215350 Email: pramodbgai@gmail.com |
|
[HTML Abstract]
[PDF Full Text]
|
| How to cite this article: |
| Goudar PHK, Joshi R, Hiremath SV, Gai PB. Mohr syndrome: A rare case of oro-facial-digital syndrome type II with congenital heart disease. International Journal of Case Reports and Images 2012;3(2):32-35. |
|
Abstract
|
|
Introduction:
Oral-Facial-Digital (OFD) syndrome is the collective name of a group of rare inherited syndromes characterised by malformations of the mouth, face, hands and feet. Currently, nine OFD syndromes have been identified and among them OFD I and OFD II are comparatively common. These two can be distinguished from each other with the help of skeletal X-rays and by the observation of various specific symptoms and also have different patterns of inheritance.
Case Report: A case report of a female child with features of OFD II also called as Mohr syndrome is presented in this study. Comparison with all the previously determined features of Mohr syndrome was done with this case and investigations were carried. Conclusion: The report showed the confirmation of Mohr Syndrome in this child along with some uncommon features like congenital heart disease. | |
|
Key Words:
OFDS-Type II, Lobulated tongue, Triplication of great toes, Typical facies, Post and preaxial polydactyly
| |
|
Introduction
| ||||||
|
Mohr syndrome or Oro-Facial-Digital syndrome type II (OFD-II) is characterised by tongue lobulation, midline cleft lip, high arched or cleft palate, broad nasal root with wide bifid nasal tip, hypertelorism, micrognathia, brachydactyly, syndactyly and polydactyly, bilateral reduplicated hallux, conductive hearing loss and normal intelligence. [1] In view of the different modes of inheritance and different prognoses of two oro-facio-digital syndromes, type 1 and type 2, it is important to establish a correct diagnosis in these patients. A female child with features of oro-facio-digital syndrome type-II is being reported in this study. | ||||||
|
Case Report
| ||||||
|
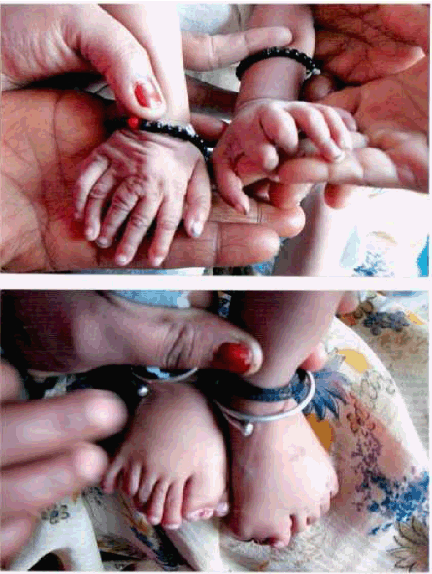
The proband, a female was the second child born to healthy, 2nd degree consanguineous, Indian parents residing at Shibargatti a rural place in Dharwad district, Karnataka, India. The first male child which also had the similar symptoms died at the age of three months, two years before the birth of proband. The normal vaginal delivery of the proband took place at 37th week in the district civil hospital of Dharwad. Birth weight of this female child was 2200 gm, which was quite less as compared to a normal healthy child's birth weight. She was unable to adjust for natural feeding due to her lobulated tongue and cleft palate. Frequent fever, tachypnea and recurrent respiratory infections were observed in later days. The child had many typical and common features of Mohr syndrome along with few uncommonly reported features. The typical findings of Mohr syndrome found in this child were: face consisting of hypertelorism, broad nasal root with wide bifid-nasal-tip. There were some unusual findings in the facies like presence of epicanthic folds and low set ears (figure 1). The typical oral features noted in the child were, lobulated tongue with papilliform protuberances, complete cleft palate with absence of uvula (figure 2A). Digital features included bilateral hallucial triplication with polysyndactyly, bilateral postaxial polydactyly of the hands, bilateral and preaxial polydactyly of the feet (figure 2B). Child had total of 31 digits. Child also had some unusual features like the presence of systolic murmur, suggestive of congenital heart disease, tachypnea [2] and recurrent respiratory infections. The congenital heart disease (CHD) was confirmed to be single atrium by Ecocardiographic studies. Few unique and few uncommon features noted in this child were low set ears, epicanthic folds, complete cleft palate, CHD (single atrium) and bilateral hallucial triplication (figure 2B). Different biochemical tests of the proband were performed at her age of nine months. The haemoglobin test by Sahli's method showed 10.5 g/dl, total WBC count was 10,800/mm3 with differential count P-70, L-29, E-01, M-00. Erythrocyte sedimentation rate (ESR) by Westergren method showed 10 mm/1st hour. CRP test was negative. Blood glucose level was 30 mg/dl and Sr., calcium was 7.6 mg/dl. Ultra sonogram of the abdomen showed normal echogenic liver. No abscess or mass lesion in the liver was identified. Gall Bladder was contracted. Common bile duct appeared normal and no calculi was seen in the duct. Pancreatic duct was not well visualised. KUB scanning and spleen tests were found to be normal. Karyotyping studies were conducted on the parents and grandmother of the proband. Elder brother of the father (proband's uncle) who has a 12-year-old healthy female child was also subjected to karyotyping studies. But the results of all the four individuals examined were normal, which clearly indicates that chromosomal abnormality was not responsible for this syndrome. | ||||||
| ||||||
| ||||||
| ||||||
|
Discussion
| ||||||
|
Orofaciodigital syndrome (OFDS) type II is a genetic condition that was first described in 1941 by Mohr. OFDS type II belongs to a group of disorders called orofaciodigital syndromes (OFDS) characterized by mouth malformations, unique facial findings, and abnormalities of the fingers and/or toes. Other organs might be affected in OFDS, defining the specific types. OFDS type II is very similar to oral-facial-digital syndrome (OFDS) type I. However, the following differences are noted, OFDI is caused by mutation in OFD I gene located on X chromosome. The involvement of skin, hair, nail, kidneys and Central Nervous System are very common. Although it is known that OFDS type II is genetically controlled as it is evident by the birth of two siblings with the same syndrome in same family, the exact gene that causes the syndrome has not been identified. The condition is believed to be inherited in an autosomal recessive pattern. Treatment is based on the symptoms present in the patient. [3] The brachydactyly characters observed in the patient, which is one of the major symptoms of Mohr syndrome is explained by Norwegian geneticist Otto L. Mohr. [4] Typically in the Mohr syndrome, the hands show postaxial polydactyly and the feet show preaxial polydactyly which are seen in the proband. [5] Along with that, the proband also showed the tachypnea and respiratory disorders as explained by Gustavson et al. [6] Congenital heart disease is an uncommon finding which is noted in the proband. [7] Unique signs like bilateral hallucial triplication, complete cleft palate with absence of uvula, lowset ears and epicanthic folds were observed in the proband which are not reported in the earlier studies (table 1, 2). By the study and detailed investigation of the proband, the reports strongly suggest the characters of Mohr syndrome. | ||||||
| ||||||
| ||||||
|
Conclusion
| ||||||
|
Here we have reported a rare case of familial Mohr syndrome i.e. OFD type II with congenital heart disease. The proband, a girl born to 2nd degree consanguineous parents not only shows all the features of Mohr syndrome but manifests some of the unusual features like bilateral hallucial triplication, lowest ears and epicanthic folds which are not reported in earlier studies. The proband also showed congenital heart disease which is rarely reported. In this sense this is a very rare and unusual case report. | ||||||
|
References
| ||||||
| ||||||





|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions:
Prakashgouda HK Goudar - Acquisition of data, Analysis and interpretation of data, Drafting the article, Final approval of the version to be published Ravindra Joshi - Analysis and interpretation of data, Drafting the article, Critical revision of the article, Final approval of the version to be published Shivaprakash V Hiremath - Analysis and interpretation of data, Drafting the article, Critical revision of the article, Final approval of the version to be published Pramod Bhimarao Gai - Conception and design, Drafting the article, Critical revision of the article, Final approval of the version to be published |
|
Guarantor of submission:
The corresponding author is the guarantor of submission. |
|
Source of support:
None |
|
Conflict of interest:
Authors declare no conflict of interest. |
|
Copyright:
© Prakashgouda HK Goudar et al. 2012; This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any means provided the original authors and original publisher are properly credited. (Please see http://www.ijcasereportsandimages.com/copyright-policy.php for more information.) |
|
|