| Table of Contents |  |
|
CASE REPORT
|
| Degradation of myofibrillar proteins and inadequate antioxidants in selective muscle wasting of limb girdle muscular dystrophy |
| Rajakumar Dhanarajan1, Anilkumar B Patil2, Mathew Alexander3, Geeta Chacko4, Anna Oommen5 |
|
1Senior Research Fellow, Section of Neurochemistry, Department of Neurological Sciences, Christian Medical College, Vellore, Tamilnadu, India.
2Assistant Professor, Section of Neurology, Department of Neurological Sciences, Christian Medical College, Vellore, Tamilnadu, India. 3Professor, Section of Neurology, Department of Neurological Sciences, Christian Medical College, Vellore, Tamilnadu, India. 4Professor, Section of Neuropathology, Department of Neurological Sciences, Christian Medical College, Vellore, Tamilnadu, India. 5Senior Scientist Grade-I, Section of Neurochemistry, Department of Neurological Sciences, Christian Medical College, Vellore, Tamilnadu, India. |
|
doi:10.5348/ijcri-2011-06-37-CR-2
|
|
Address correspondence to: Dr. Rajakumar Dhanarajan Neurochemistry Laboratory Department of Neurological Sciences Christian Medical College Vellore 632 004 India Phone: 91-9790570526 Fax: 91-416-2232035 Email: dhanurajan@hotmail.com |
|
[HTML Abstract]
[PDF Full Text]
|
| How to cite this article: |
| Dhanarajan R, Patil AB, Alexander M, Chacko G, Oommen A. Degradation of myofibrillar proteins and inadequate antioxidants in selective muscle wasting of limb girdle muscular dystrophy. International Journal of Case Reports and Images 2011;2(6):6-11. |
|
Abstract
|
|
Introduction:
An unexplained feature of inherited muscular dystrophies is the wasting of selective muscle. Transcriptional signatures that differ between muscles may contribute to selective muscle wasting of muscular dystrophies. Biochemical signaling pathways involved in selective wasting have not been studied.
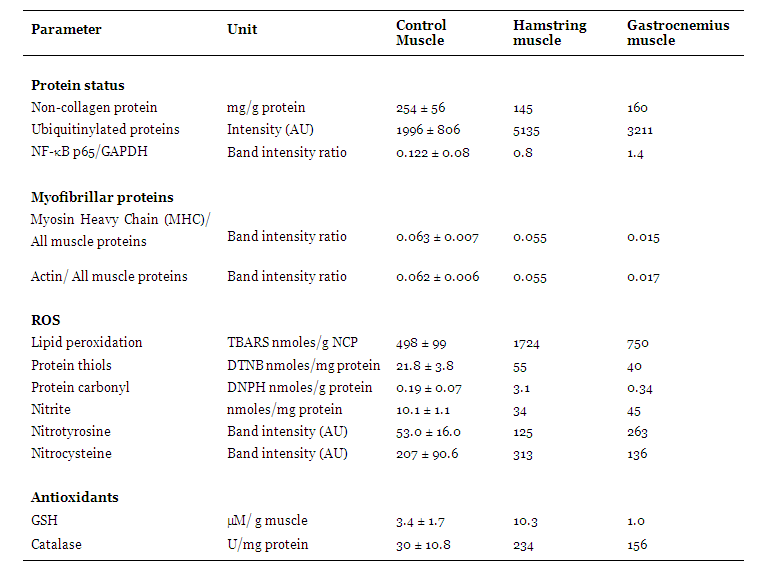
Case Report: This study examined signaling mechanisms that stimulate protein degradation, which may govern selective muscle wasting, in a patient with Miyoshi myopathy. Oxidative stress, nitric oxide signaling and protein degradation through the ubiquitin proteasome pathway were studied in relatively spared hamstring and wasted gastrocnemius muscle of the patient and in control muscle. Oxidative stress occurred in spared and wasted Miyoshi myopathic muscle. A strong anti-oxidant response was observed in spared muscle. Tyrosine nitration of 56kDa protein(s) was 2.3 and 5 fold higher in spared and wasted muscles respectively compared to normal. Nitrocysteinylated proteins were comparable between spared hamstring and normal muscle but reduced 35% in gastrocnemius muscle compared to normal. Ubiquitinylated proteins were increased 2.6 fold in the hamstring muscle and 1.6 fold in the gastrocnemius muscle compared to normal. The protein content of the hamstring and gastrocnemius muscles was reduced 43% and 37% respectively compared to normal. Myosin heavy chain and actin levels were normal in hamstring muscle but reduced nearly four fold in gastrocnemius muscle compared to normal. Conclusion: Oxidative and nitrosative stress and loss of actin and myosin were associated with selective muscle wasting of Miyoshi myopathy. A strong anti-oxidant response that protects myofibrillar proteins against degradation may spare muscle in Miyoshi myopathy. | |
|
Key Words:
Actin, Miyoshi myopathy, Myosin, Nitric oxide, Ubiquitin-proteasome pathway
| |
|
Introduction
| ||||||
|
An intriguing feature of muscular dystrophies is the involvement of selective tissue with disease progression despite a common mutation in all tissues. Transcriptional signatures that differ between muscles suggest that specific signaling pathways may be associated with selective muscle wasting of muscular dystrophies. [1] The proteomics of these signaling pathways have been not explored in detail. Limb girdle muscular dystrophies (LGMD) are among the most common muscular dystrophies which target the limb girdle musculature. Dysferlinopathies, autosomal recessive LGMD, are due to absence or reduction of the muscle membrane protein, dysferlin. [2] A case of dysferlinopathy is presented to highlight preferential loss of specific proteins in selectivity of genetic disease. The major muscle proteins myosin and actin, oxidative and nitrosative stress that activate protein degradation and the ubiquitin proteasome protein-degrading pathway, were assessed in relatively spared hamstring muscle and wasted gastrocnemius muscle of a patient with Miyoshi myopathy, to determine their role in selective muscle dystrophy.
| ||||||
|
Case Report
| ||||||
|
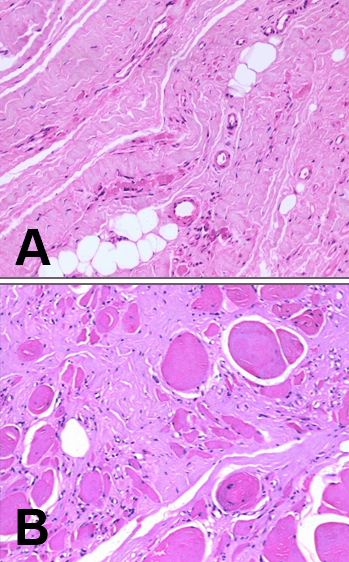
The patient was a 22-year-old male who presented with a 4-year history of pain in his calf, thigh and gluteal muscles while walking. He developed thinning and weakness of calf muscles and later of the thigh muscles. He is born of a non-consangenious union and has no family history. Neurological examination revealed marked bilateral wasting of the calf muscles, foot muscles, hamstring and quadriceps. Lower limbs had marked weakness of the plantar flexion of the ankle (MRC 3/5) and the other groups of muscles had an MRC 4/5. Deep tendon reflexes were absent at the ankles. He had a waddling gait and could not walk on his toes. His serum creatine kinase was 9431 units/litre and the EMG was supportive of a myopathic process. He underwent a muscle biopsy of his right gastrocnemius muscle. Six months later MR imaging of the lower limbs led to a muscle biopsy of the left hamstring muscle. The muscle biopsies were subject to histology, immunohistochemistry and western blot analysis. Normal muscles obtained from five patients undergoing surgery for clubfoot correction served as controls. All muscle samples were obtained with informed consent. Methods Histology: SDS-polyacrylamide gel electrophoresis (SDS-PAGE) Western blots Oxidative and nitrosative stress parameters Diagnosis Although mutation analysis is the gold standard for diagnosis of dysferlinopathy, clinical features specific to dysferlinopathy with immune blots negative for dysferlin are considered diagnostically adequate and comparable to mutational analysis. [3] The patient in this study was diagnosed as Miyoshi myopathy based on clinical findings, muscle histopathology, immunohistochemistry and western blots negative for dysferlin. Results Histopathology, immunohistochemistry and western blots | ||||||
|
| ||||||
|
| ||||||
|
Muscle proteins Oxidative and nitrosative stress Antioxidants in muscles | ||||||
| ||||||
|
| ||||||
|
| ||||||
|
Discussion
| ||||||
|
The arguments in this study for suggesting mechanisms of selective and preferential loss of the gastrocnemius muscle in Miyoshi myopathy are based on the earlier severe wasting of this muscle compared to the hamstring muscle that remains preserved even six months later. The absence of dysferlin was associated with significant protein loss by degradation through the ubiquitin proteasome system in both muscles. The nature of the lost proteins were different and this maybe important in the pathogenesis of Miyoshi myopathy. Loss of actin and myosin in the gastrocnemius muscle resulting in a 40% decrease in the myosin to actin ratio may account for weakness of this muscle in Miyoshi myopathy. Muscles with the capacity to protect their major myofibrillar proteins from degradation maybe spared in Miyoshi myopathy, as noted in the hamstring muscle, where loss may mainly be of sarcoplasmic and extracellular proteins. Mediators of intracellular signaling that regulate protein degradation include oxidative and nitrosative stress. [4] [5] [6] Absence of dysferlin appears to induce oxidative stress in both spared hamstring and affected gastrocnemius muscles. The ability to mount a strong anti-oxidant defense in the spared muscle but only a weak response in the wasted muscle suggests that guarding against oxidative damage is important to spare muscle in Miyoshi myopathy. In the oxidative environment of the muscle, NO can convert to reactive nitrogen species which can modify proteins and activate different signaling pathways including those that act through NF-κB. [7] Posttranslational modifications induced by NO, of tyrosine nitration and S-nitrocysteinylation, can be protective or induce protein degradation and cell death. [8] The NO profiles of spared and affected muscles of Miyoshi myopathy suggest a role for NO in committing cells to death. In muscles that are spared, although NO is elevated, the NO signature resembles that of normal muscle with respect to nitro-cysteinylated proteins. Tyrosine nitration of protein(s) of 56kDa is minor. Elevated levels of GSH in this muscle may scavenge NO and maintain the normal NO profile. In contrast predominant tyrosine nitration of 56kDa protein(s) with concomitant reduction of nitro-cysteinylated proteins and increased NF-κBp65 in wasted muscle may underlie signaling pathways that direct selective muscle wasting of Miyoshi myopathy. Similar findings are noted in disuse-induced atrophy. In rats subject to space flights or hind limb suspension, where the gastrocnemius muscle is selectively wasted, preferential wasting of myosin heavy chain, increased lipid peroxidation, decreased reduced glutathione and increased muscle protein polyubiquitinylation are also reported. [9] [10]
| ||||||
|
Conclusion
| ||||||
|
Oxidative and nitrosative stress that lead to nitration of tyrosine on protein(s) of 56kDa may signal degradation of the actinomyosin complex and result in weakness. A strong antioxidant defense that guards against loss of contractile proteins may spare the muscle.
| ||||||
|
References
| ||||||
| ||||||



| [HTML Abstract] [PDF Full Text] |
|
Acknowledgement:
R. Dhanarajan is supported by a senior research fellowship of the Indian Council of Medical Research (Project No 5/4 -5/2/Neuro/2008). |
|
Author Contributions:
Rajakumar Dhanarajan - Conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Critical revision of the article, Final approval of the version to be published Anilkumar B Patil - Conception and design, Drafting the article, Critical revision of the article, Final approval of the version to be published Mathew Alexader - Conception and design, Drafting the article, Critical revision of the article, Final approval of the version to be published Geeta Chacko - Conception and design, Drafting the article, Critical revision of the article, Final approval of the version to be published Anna Oommen - Conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Critical revision of the article, Final approval of the version to be published |
|
Guarantor of submission:
The corresponding author is the guarantor of submission. |
|
Source of support:
None |
|
Conflict of interest:
The author(s) declare no conflict of interests |
|
Copyright:
© Rajakumar Dhanarajan et al. 2011; This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any means provided the original authors and original publisher are properly credited. (Please see Copyright Policy for more information.) |
|
|