| |
|
|
|
Case Report
| ||||||
| Unmasking IgG4-related autoimmune pancreatitis from pancreatic cancer: A lesson learned | ||||||
| Ashwad Afzal1, Seema Chittalae1, Ivan Wong1, Petros Efthimiou1 | ||||||
|
1MD, Department of Internal Medicine, New York Methodist Hospital affiliate of Weill Medical College of Cornell University, Brooklyn, New York, USA.
| ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article]
[Similar article in Pumed] [Similar article in Google Scholar] 
|
| How to cite this article |
| Afzal A, Chittalae S, Wong I, Efthimiou P. Unmasking IgG4-related autoimmune pancreatitis from pancreatic cancer: A lesson learned. Int J Case Rep Images 2016;7(2):123–126. |
|
Abstract
|
|
Introduction:
Autoimmune pancreatitis (AIP) is difficult to distinguish from pancreatic cancer. An AIP has a similar clinical presentation and is one component of a systemic disease, Immunoglobulin G4 (IgG4) related sclerosing disease. IgG4 related sclerosing disease is characterized by extensive IgG4 positive plasma cells and T-lymphocytes that may involve the pancreas and other organs.
Case Report: A 77-year-old male presented with nausea, vomiting, decreased appetite, and 39 lbs weight loss over three months. An endoscopic ultrasound showed one 3.3x2.7 cm mass on the tail of the pancreas extending towards the splenic hilum with no lymph node involvement. A fine needle aspiration was indeterminate. The patient underwent distal pancreatectomy and splenectomy for presumed pancreatic cancer, however, the pathology report was negative for malignancy. Instead, the microscopic examination revealed dense lymphoplasmacytic infiltrate staining positive for IgG4, onion-skin pattern fibrosis, and lobular atrophy in the medium sized pancreatic duct that was consistent with AIP. Conclusion: IgG4 related sclerosing disease is a systemic disorder that may involve multiple organs. The clinical manifestations are similar to that of pancreatic malignancy. For this reason, diagnostic evaluation to differentiate the two disease entities is crucial, as treatments differ significantly. Treatment for AIP consists of early initiation with glucocorticoid therapy. However, poor response to treatment usually indicates advanced fibrotic changes or an alternative diagnosis. Failure to differentiate autoimmune pancreatitis from pancreatic cancer can lead to unnecessary surgery of the pancreas along with complications associated with surgery. | |
|
Keywords:
Autoimmune pancreatitis, IgG4 related sclerosing disease, Pancreatic cancer
| |
|
Introduction
| ||||||
|
Autoimmune pancreatitis (AIP) is difficult to distinguish from pancreatic cancer. They both share similar clinical presentation including an insidious with anorexia, nausea, vomiting, decreased appetite, epigastric pain, and significant weight loss. The AIP also has a similar clinical presentation and is one component of a systemic disease, immunoglobulin G4 (IgG4) related sclerosing disease. IgG4 related sclerosing disease is characterized by extensive IgG4 positive plasma cells and T lymphocytes that may involve the pancreas, bile duct, retroperitoneum, salivary glands and other organs. We illustrate a case of autoimmune pancreatitis presenting as pancreatic cancer. | ||||||
|
Case Report
| ||||||
|
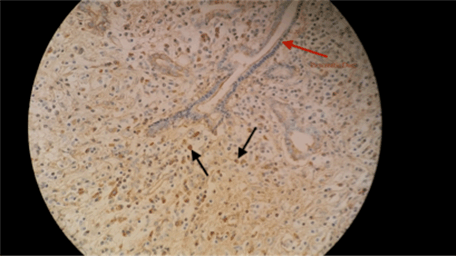
A 77-year-old legally blind male with a past medical history of hypertension, hyperlipidemia, coronary artery disease with stents, and diabetes mellitus type 2 (established for 25 years) presented with nausea, vomiting, decreased appetite, and 39 lbs weight loss over a 3-month period. He was an ex-smoker with a 30-pack-year history. There was no significant surgical or family history. His vital signs were stable and his body mass index was 24.91 kg/m2. The pertinent physical examination revealed mild tenderness to palpation over the epigastric area with no scleral icterus, and no lymphadenopathy. Routine blood work including complete blood count, renal and liver function tests were normal. An endoscopic ultrasound showed a 3.3x2.7 cm mass on the tail of the pancreas extending towards the splenic hilum with no lymph node involvement (Figure 1). A fine needle aspiration was indeterminate. The patient underwent distal pancreatectomy and splenectomy for high suspicion of pancreatic malignancy, however, the surgical pathology report was negative for malignancy. Microscopic examination revealed dense lymphoplasmacytic infiltrate with onion-skin pattern fibrosis and lobular atrophy in medium sized pancreatic ducts that was consistent with lymphoplasmacytic sclerosing pancreatitis (Figure 2). Further immunohistochemical staining showed 30–40 IgG4 positive plasma cells on high power field and labs revealed an elevated IgG4 level of 272.5 mg/dl, consistent with the diagnosis of AIP (Figure 3). | ||||||
| ||||||
| ||||||
| ||||||
|
Discussion
| ||||||
|
IgG4 related sclerosing disease is a systemic disorder that may involve multiple organs. The clinical manifestations may include anorexia, weight loss, nausea, and painless jaundice similar to that of pancreatic malignancy. For this reason, diagnostic evaluation to differentiate the two disease entities is crucial, as treatment differ significantly. In AIP, serum IgG4 levels are elevated, and levels >2 times the upper limit of normal is highly suggestive of the disease [1]. However, this may also be seen in pancreatic malignancy and thus cannot be used to differentiate the two [2]. Type 1 AIP (lymphoplasmacytic sclerosing pancreatitis) has a greater prevalence of having elevated serum IgG4 levels and a higher relapse rate compared to Type 2 AIP (idiopathic duct centric pancreatitis)[3]. There are various imaging modalities which can be used to evaluate the pancreas including computed tomography, or magnetic resonance imaging identifying a diffusely enlarged pancreas resembling a "sausage-like" appearance or a focal mass, such as in this case. Endoscopic ultrasound may reveal diffuse hypoechoic pancreatic enlargement and bile duct wall thickening. A core needle biopsy is often required for histological evaluation as opposed to a fine needle aspiration, which may be inadequate, as was portrayed in this case. The presence of lymphoplasmacytic tissue infiltration characterized by extensive IgG4 positive plasma cells and T lymphocytes deposition, fibrosis, and obliterative phlebitis on histology is diagnostic for AIP [4] [5]. Response to steroid therapy also supports the diagnosis [6]. Treatment consists of early initiation with glucocorticoid therapy [7]. A study by Matsubayashi et al. showed steroid response in 86% of patients within two weeks and in 97% of patients within four weeks by demonstrating a decrease in tumor size on abdominal ultrasound [6]. Steroid response was seen in almost 98% of patients with AIP in a study by Kamisawa et al. [8]. For resistant cases, B cell depleting agents such as Rituximab has been used with success [9]. A poor response to treatment usually indicates either advanced fibrotic changes or an alternative diagnosis. | ||||||
|
Conclusion
| ||||||
|
Autoimmune pancreatitis (AIP) is a difficult diagnosis to differentiate from pancreatic cancer, as the presentation is very similar. Clinical suspicion with laboratory markers and imaging may help aid in the diagnosis. However, laboratory markers may be normal and a biopsy is required to make the diagnosis. Failure to differentiate AIP from pancreatic cancer can lead to unnecessary resection of the pancreas along with complications of surgery, as autoimmune pancreatitis responds well to steroid therapy. We recommend patients with pancreatic mass be evaluated for AIP to avoid invasive treatment. | ||||||
|
Acknowledgements
| ||||||
|
We are thankful to Arpita Bose and the library staff at New York Methodist hospital for their dedication in helping residents with research. | ||||||
|
References
| ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions
Ashwad Afzal – Substantial contribution to conception and design, Acquisition of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Seema Chittalae – Substantial contribution to conception and design, Acquisition of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Ivan Wong – Substantial contribution to conception and design, Acquisition of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published. Petros Efthimiou – Substantial contribution to conception and design, Acquisition of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published. |
|
Guarantor of submission
The corresponding author is the guarantor of submission. |
|
Source of support
None |
|
Conflict of interest
Authors declare no conflict of interest. |
|
Copyright
© 2016 Ashwad Afzald et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information. |
|
|